CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
Abstract
Ferroptosis is an iron-dependent cell death pathway that, until recently, has been considered to be dependent on autophagy. However, recent studies have reported conflicting results, raising the question about which cell contexts determine the roles of autophagy in ferroptosis. This opinion article addresses this question by summarizing the contexts and/or diseases in which autophagy is a driver or suppressor of ferroptosis. The execution of ferroptosis depends on levels of (labile) iron, unsaturated (phospho)lipids and free radicals. We propose that the cell context in which these three factors and/or their upstream pathways are differentially regulated dictates whether autophagy positively or negatively regulates ferroptosis.
摘要
本文讨论了自噬在铁死亡(ferroptosis)中的作用。铁死亡是一种依赖铁的细胞死亡方式。传统上认为铁死亡依赖于自噬,但最近的研究结果却与这种认识存在冲突。文章旨在探讨自噬在铁死亡中作为驱动因素或抑制因素的具体细胞环境和疾病背景。文章提出,铁、不饱和(磷)脂质和自由基的水平,以及它们的上游通路,决定了自噬是促进还是抑制铁死亡。
这篇文章的核心内容是探讨自噬(autophagy)在铁死亡(ferroptosis)中的作用,以及在不同细胞环境中自噬如何调节铁死亡。文章详细总结了自噬依赖性和自噬非依赖性铁死亡的机制,并讨论了铁死亡的调控因素和潜在的治疗靶点。
内容总结
铁死亡(Ferroptosis):一种依赖铁的细胞死亡方式,由多种刺激引起,导致(磷)脂质过氧化物的积累,与凋亡和其他类型的细胞死亡不同。
自噬(Autophagy):一种细胞内部降解机制,通过溶酶体降解细胞质内容物,包括受损的细胞器和蛋白质聚集体。
研究方法
文章通过文献综述的方式,总结了自噬在铁死亡中的作用。研究方法包括:
分析自噬和铁死亡的关键调节因子,如谷胱甘肽过氧化物酶4(GPX4)、SLC7A11、ACSL4等。
探讨自噬如何通过调节铁水平、抗氧化酶活性和脂质代谢来影响铁死亡。
评估自噬在不同细胞类型和疾病模型中对铁死亡的影响。
实验结果与关键结论
铁死亡的调控因素
铁死亡依赖于铁、不饱和(磷)脂质和自由基的水平。
这些因素的上游通路在不同细胞环境中被差异性调节,决定了自噬对铁死亡的正向或负向调节作用。
自噬依赖性铁死亡
自噬通过降解铁蛋白(ferritin)增加细胞内游离铁水平,从而促进铁死亡。
自噬还通过调节GPX4、SLC7A11等关键因子的活性或表达来影响铁死亡。
例如,在RSL3处理的PANC1细胞中,GPX4通过TMEM164/ATG5依赖的非典型自噬途径被降解。
自噬非依赖性铁死亡
某些情况下,铁死亡可以不依赖于自噬发生。例如,WIPI4的缺失导致自噬独立的铁死亡,通过影响脂质代谢促进铁死亡。
溶酶体功能和完整性在铁死亡中起重要作用,即使在自噬缺陷的细胞中,溶酶体也可以通过非典型途径调节铁死亡。
自噬与铁死亡的共存
自噬和铁死亡可能共享上游信号通路,例如mTORC1抑制可以同时促进自噬和铁死亡。
p53在自噬和铁死亡中具有双重作用,通过调节脂质代谢影响铁死亡。
细胞类型和病理环境的影响
自噬对铁死亡的影响可能因细胞类型和病理环境而异。例如,在某些癌症细胞中,自噬可能通过降解转铁蛋白受体1(TfR1)来抑制铁死亡,而在其他情况下,自噬可能通过清除产生ROS的线粒体来缓冲铁死亡。
研究意义
文章强调了自噬在铁死亡中的复杂作用,并指出了自噬依赖性和非依赖性铁死亡的机制。这些发现对于理解铁死亡的调控机制以及开发针对铁死亡的治疗策略具有重要意义。特别是,文章提出了自噬和铁死亡之间的相互作用可能因细胞类型和病理环境而异,这为未来的细胞生物学研究和临床应用提供了新的视角。
未来研究方向
文章提出了几个未来研究的方向,包括:
自噬基因(包括ATGs)是否通过其自噬或非自噬功能调节铁死亡。
在自噬途径的不同阶段抑制自噬对铁死亡的影响。
自噬如何影响铁死亡的起始因素与促进细胞死亡的因素。
确定决定不同铁死亡途径自噬依赖性的组织特异性因素。
图文摘要
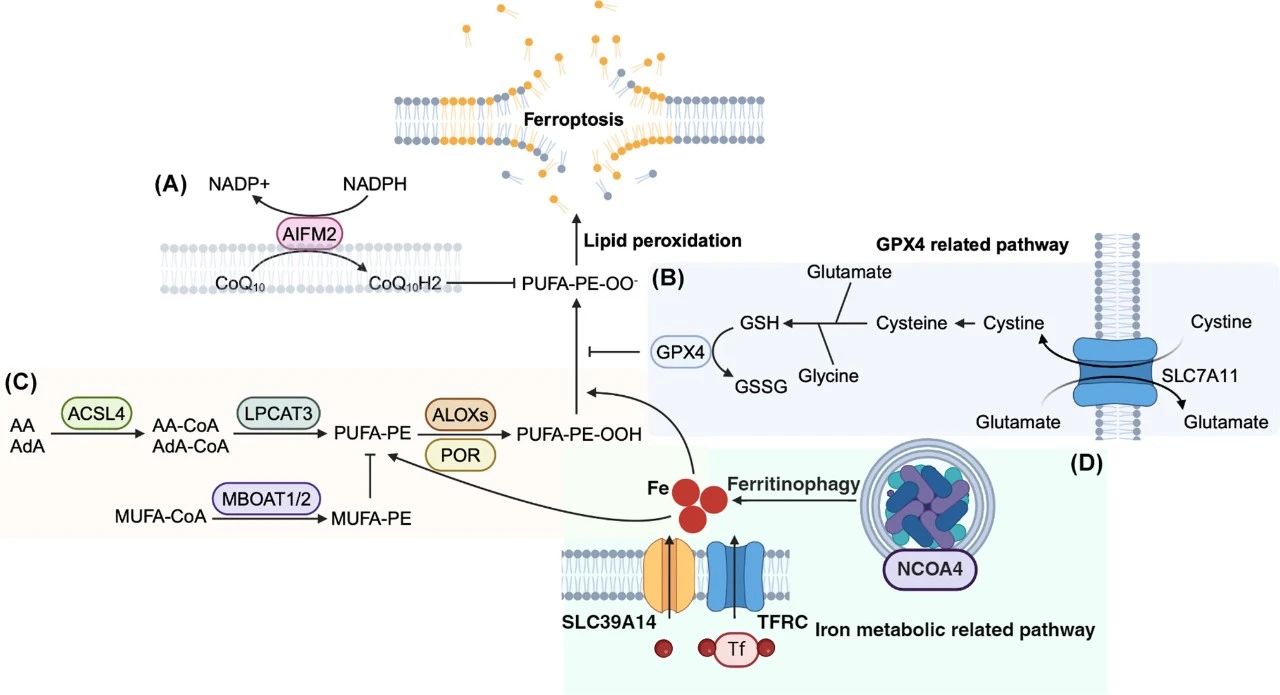
图1. Ferroptosis(铁死亡)
细胞内游离铁或含铁酶与富含多不饱和脂肪酸(PUFA)的磷脂相互作用,特别是磷脂酰乙醇胺(PE),导致膜脂过氧化物(PUFA-PE-OO)水平升高,进而导致膜破裂。这一过程可以通过谷胱甘肽过氧化物酶4(GPX4)和凋亡诱导因子线粒体相关2(AIFM2)系统来限制。
(A)AIFM2/FSP1通过NAD(P)H产生还原型辅酶Q10(CoQ10H2)或维生素K。还原型抗氧化剂可以向磷脂过氧自由基(PUFA-PE-OO·)捐赠电子,终止脂质过氧化。
(B)GPX4是抵御脂质过氧化的主要防御机制之一,需要由半胱氨酸、甘氨酸和谷氨酸合成的谷胱甘肽(GSH)。半胱氨酸由系统xc-导入,其转运单元由SLC7A11编码。
(C)ACSL4催化PUFAs(花生四烯酸(AA)、肾上腺酸(AdA))与辅酶A的酯化,PUFA-CoA可以被纳入磷脂(PUFA-PE)中。溶血磷脂可以被LPCAT3用酰基辅酶A重新酰化,从而重新形成磷脂。磷脂过氧化物(PUFA-PE-OOH)可以通过酶促反应(通过脂氧合酶(ALOXs)或P450氧化还原酶(POR))和非酶促自由基介导反应生成。膜结合O-酰基转移酶结构域1/2(MBOAT1/2)增加单不饱和脂肪酸(MUFAs)的合成,通过磷脂重塑抑制铁死亡。
(D)Fe3+-转铁蛋白复合物通过转铁蛋白受体(TFRC)或SLC39A14摄取,并与铁蛋白复合物储存,铁蛋白复合物可以通过NCOA4介导的铁蛋白降解,释放游离铁进入细胞质。
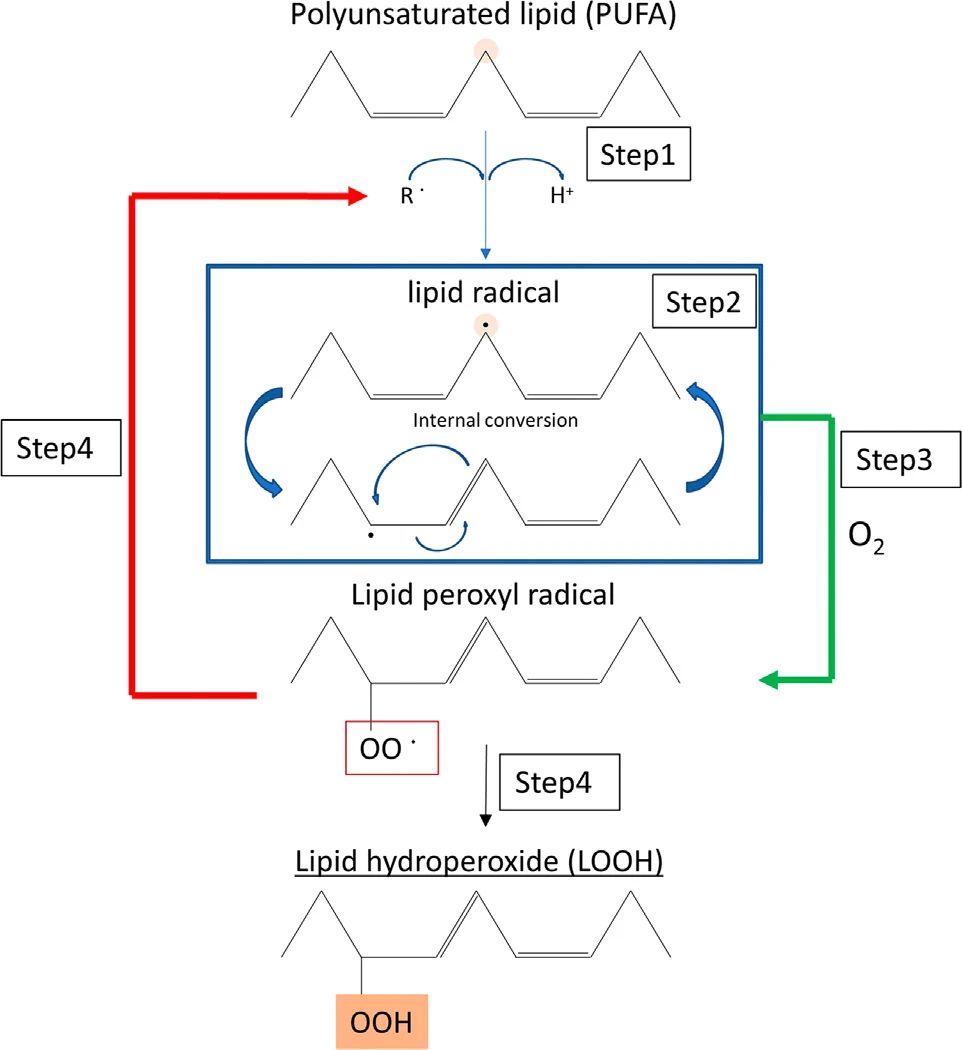
图2. 脂质过氧化
自由基提取多不饱和脂肪酸(PUFAs)中两个碳-碳双键夹着的烯丙基氢(步骤1)。碳自由基迅速转化为共轭二烯(步骤2)。在传播步骤中,这些脂质自由基与氧气反应生成脂质过氧自由基(步骤3)。在步骤4中,这些自由基可以从另一个脂质分子中抽象一个氢原子,生成下一个过氧自由基(链反应)(红色箭头)和脂质过氧化氢(绿色箭头)。
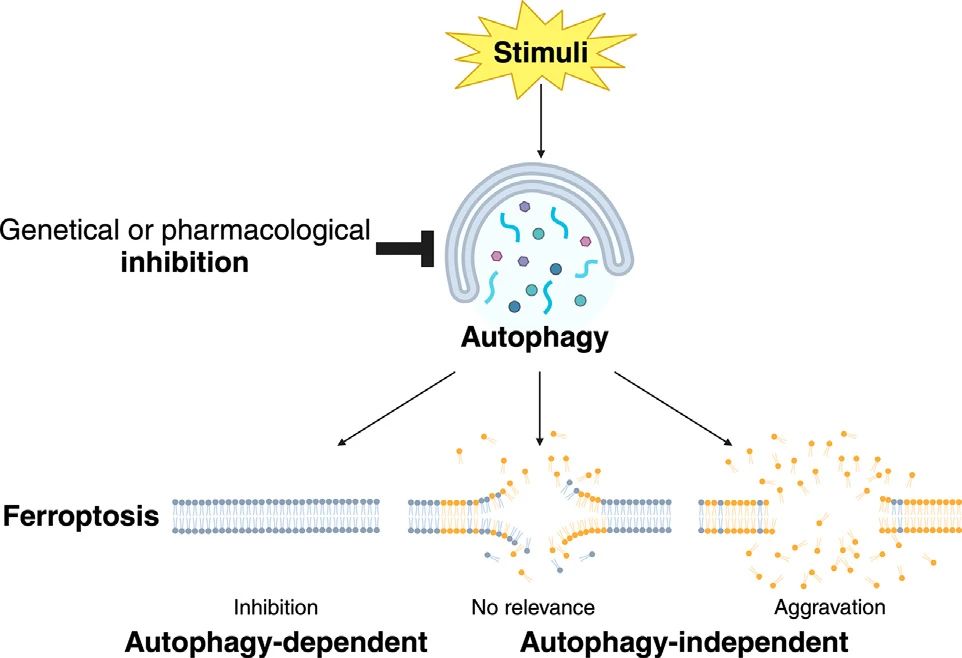
图3. 铁死亡的刺激或抑制的影响因素
铁死亡可以被自噬刺激或抑制,或者与自噬无关,这取决于细胞环境。一些研究表明自噬阻断只能部分抑制铁死亡。其他研究表明自噬阻断并不能减轻甚至促进了铁死亡。这些差异可能是由于细胞类型、铁死亡诱导剂的处理时间和浓度不同。
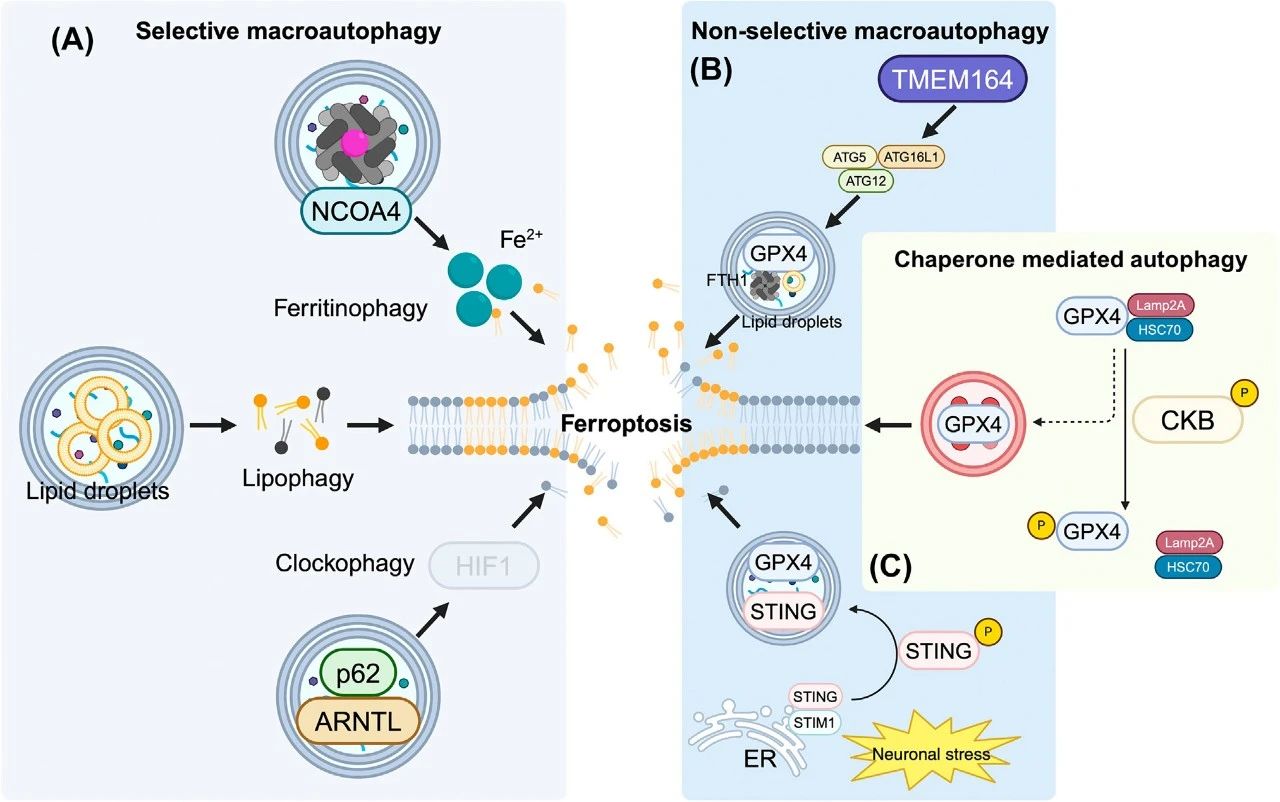
图4. 自噬依赖性铁死亡
(A)选择性巨自噬。核受体共激活因子4(NCOA4)介导的铁蛋白降解增加了细胞质中的游离铁池(Fe2+)。脂质滴自噬通过减少细胞的脂质储存能力来增强细胞对铁死亡的敏感性。通过选择性降解芳烃受体核转位蛋白样蛋白(ARNTL)来抑制HIF1,降低了多不饱和脂肪酸(PUFAs)的可用性,从而促进铁死亡。
(B)非选择性巨自噬。谷胱甘肽过氧化物酶4(GPX4)、铁蛋白重链1(FTH1)和脂质滴在依赖TMEM164/ATG5的方式下被降解。神经元应激导致磷酸化干扰素刺激基因(STING)解离,随后通过WIPI2依赖性自噬被降解。
(C)GPX4也可以通过分子伴侣介导的自噬被降解,而不需要肌酸激酶的磷酸化,如右图所示。
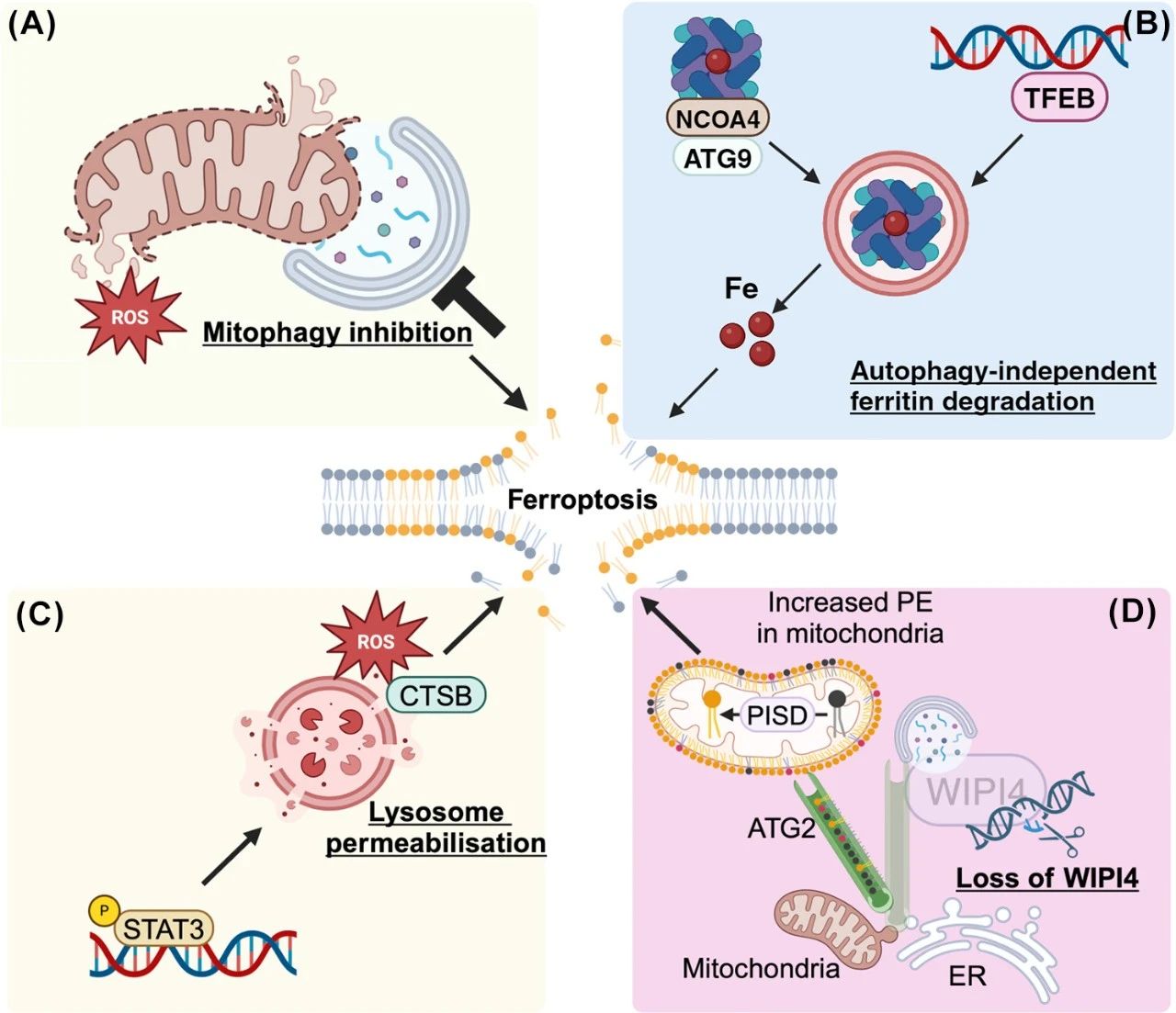
图5. 自噬独立的铁死亡
(A)线粒体自噬可以通过清除产生活性氧(ROS)的线粒体来保护细胞免受铁死亡。然而,线粒体在铁死亡中的不同作用可能决定了线粒体自噬是保护性的还是有害的。
(B)核受体共激活因子4(NCOA4)-铁蛋白复合物可以通过Unc-51样激酶(ULK)1/2-FIP200和ATG9的非自噬功能被靶向到溶酶体。TFEB核转位通过溶酶体酸化和转铁蛋白受体1(TfR1)的合成促进铁死亡。TfR1是细胞内铁摄取和ROS生成以及铁死亡所必需的。
(C)信号转导和转录激活因子3(STAT3)介导的组织蛋白酶B和L的表达和释放通过促进溶酶体膜的通透性来促进铁死亡。
(D)在β-螺旋桨蛋白相关神经退行性病变中,WIPI4功能丧失使ATG2从自噬体转移到内质网-线粒体接触位点,从而促进磷脂酰丝氨酸从内质网向线粒体的转运,然后在线粒体中被转化为磷脂酰乙醇胺。PE水平的增加促进了脂质过氧化,推动了铁死亡。
参考文献:
Zhu Y, Fujimaki M, Rubinsztein DC. Autophagy-dependent versus autophagy-independent ferroptosis. Trends Cell Biol. 2025 Mar 5:S0962-8924(25)00005-4. doi: 10.1016/j.tcb.2025.01.005. Epub ahead of print. PMID: 40050185.

