CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
《Science》近期发表的一项研究提出,扩张型心肌病的持续恶化,并不只是心肌细胞收缩功能下降的直接结果,心脏成纤维细胞对异常机械环境的放大反应,同样是推动病程进展的重要环节。研究者认为,当原发性的肌节缺陷削弱心肌收缩后,心脏微环境中的被动力学状态会随之改变,而成纤维细胞能够感知这类变化,并进一步重塑细胞外基质,最终加重心脏扩张和功能损害。
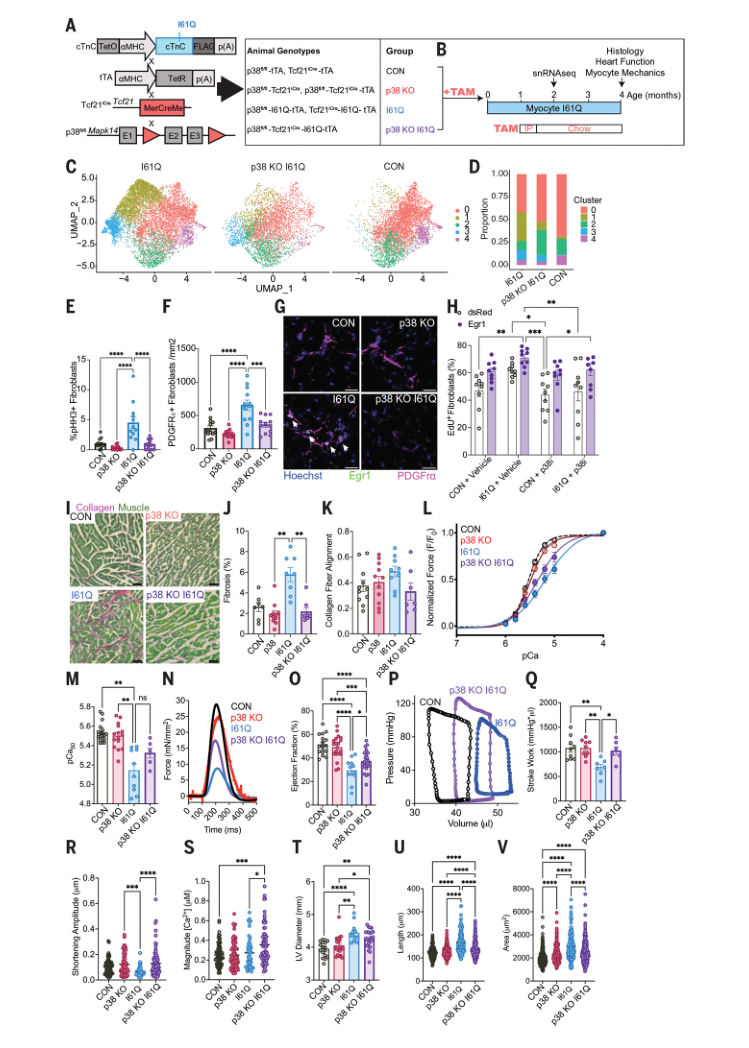
成纤维细胞特异性p38缺失可纠正I61QcTnC转基因小鼠的心脏扩张和收缩功能障
长期以来,扩张型心肌病的解释主要集中在“肌节异常—收缩障碍—心室扩张”这条主线,因此治疗思路也多围绕改善心肌细胞功能展开。但这项研究把视角推进到了心肌微环境。作者发现,在遗传性扩张型心肌病模型中,成纤维细胞的异常激活出现在明显心脏扩张和广泛纤维化之前,提示它们并非单纯对损伤作出被动反应,而可能更早参与了疾病重构过程。
在I61Q-cTnC转基因小鼠中,研究者观察到,心脏在尚未出现典型偏心性肥大前,就已经表现出被动刚度增加和胶原纤维排列异常。进一步的单核转录组分析显示,具有机械转导和高增殖特征的成纤维细胞亚群明显扩增,随着心肌收缩减弱,最先被重新编程的,不仅是心肌细胞本身,还包括负责维持基质结构的成纤维细胞。
机制上,研究进一步指出,成纤维细胞主要是通过细胞外基质与受体之间的相互作用,感知异常的舒张期机械环境。基质筛选、共培养和张力传感实验显示,Ⅳ型和Ⅵ型胶原相关信号可能在其中发挥关键作用,而p38、YAP等机械转导节点参与了后续的增殖与重塑程序。这个过程带来的直接后果,是成纤维细胞增殖增加、基质重排增强、组织逐渐僵硬,进而把原本来自心肌细胞的功能缺陷放大为更严重的整体心脏重构。
更关键的是,这项研究并没有停留在相关性描述。作者构建了成纤维细胞特异性p38缺失模型,结果发现,在阻断这一通路后,小鼠的心脏扩张程度减轻,收缩功能也得到改善。这说明成纤维细胞的机械应答并非疾病进展中的伴随现象,而是具有因果意义的致病环节,它们可能从更早阶段就参与决定了扩张型心肌病的严重程度。
这项工作的意义,在于它重新界定了扩张型心肌病中“谁在推动病程”。过去被更多视作结果的纤维化和基质重塑,在这里被提前到了病程前端,成为可以干预的关键过程。这也提示未来治疗策略或许不能只盯着心肌细胞本身,还应关注成纤维细胞及其介导的机械信号放大效应,尤其是围绕p38相关通路的精准调控。
doi:10.1126/science.adv9157.

