CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
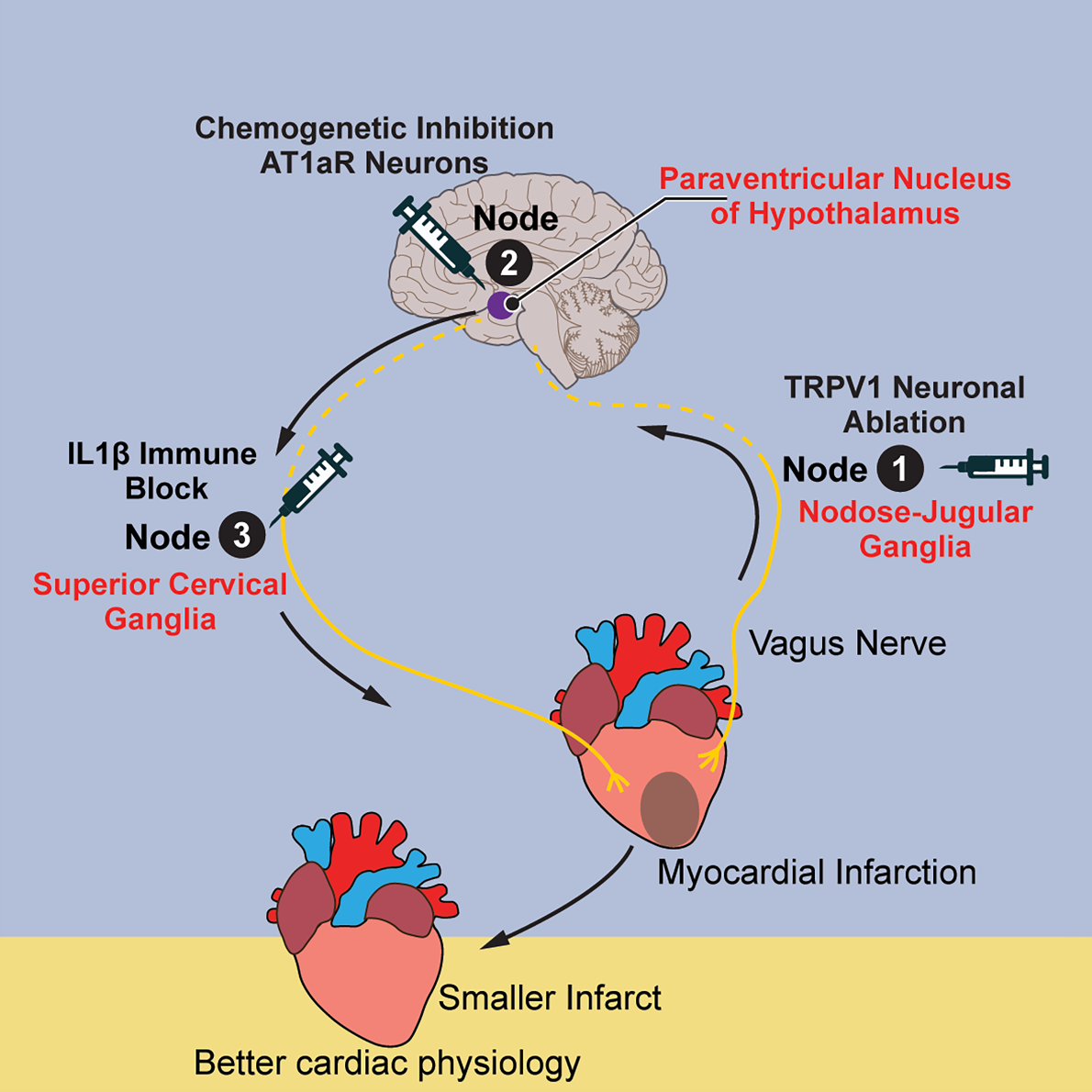
这篇发表于《Cell》的研究提出,心肌梗死后存在一条由TRPV1阳性迷走感觉神经元、下丘脑室旁核AT1aR神经元和颈上神经节IL-1β信号串联而成的“三节点”心-脑-免疫环路。在小鼠模型中,对其中任一节点进行干预,都能改善梗死后的心功能和组织损伤。这项工作值得看,不只是因为补上了一条更完整的“心脑轴”机制路径,更因为它把神经活动、炎症放大和边缘区损伤扩展放进了同一框架里,直接指向了抗重构干预的新入口。
心肌梗死后的发生的电重构、交感过度激活、炎症持续和心室重构,是推动心功能恶化甚至心衰的重要过程。既往解释更多聚焦于局部坏死、循环炎症因子和经典交感兴奋,但哪些外周感觉输入把梗死信号送入中枢,又如何被转译成持续的神经免疫损伤,长期缺少完整机制链。本文把迷走感觉输入引入这一问题,并把中枢整合节点和交感外周效应节点接上,给出的不再是单点解释,而是一条跨器官、跨细胞类型的连续路径。
研究首先把关键变化定位在心脏梗死后的边缘区。作者通过结节-颈静脉神经节单细胞测序、病毒示踪、免疫染色以及组织透明化联合光片成像,鉴定到一群TRPV1阳性迷走感觉神经元在心梗后增加,并且其神经纤维沿心室后向前轴包绕并增生于borderzone。这里的传递方式不是代谢物扩散或囊泡运输,而是感觉神经输入及其后续神经传导。功能实验显示,消融这群神经元后,心梗小鼠的QRS异常减轻,射血分数、左室容积和内径改善,同时TH阳性交感纤维减少,而Ki67、CD31和VEGF升高。这说明边缘区并非被动承受损伤,而是在神经与炎症的共同作用下持续被重塑。
作者随后把这条路径继续往中枢和外周交感端延伸。研究发现,心梗后下丘脑室旁核内cFos升高,而且被激活的神经元与AT1aR共定位;抑制PVN内AT1aR神经元后,表型基本复制了TRPV1迷走感觉神经元消融的效应,提示梗死信号被送到下丘脑后,会被转译成持续的交感输出。再往下游,作者用心肌逆行示踪和SCG定向AAV示踪证明,颈上神经节确实直接支配心脏,而且心梗后其神经投射增强,同时SCG内IL-1β升高。到这里,信号方向已经比较清楚:TRPV1迷走感觉神经元将损伤信息送到PVN的AT1aR神经元,随后中枢通过交感通路影响SCG,SCG再把高交感和局部神经炎症效应反馈到心脏边缘区。单核转录组和空间转录组进一步显示,干预后边缘区与远端区的基因程序转向收缩调控、细胞连接装配、血液循环和抗氧化防御,同时炎症相关基因下调,对应的组织层面表型则是梗死扩展受限、边缘区保护增强以及心功能恢复。
这项研究的强项是因果链条比较完整,但人群层面的临床证据仍然偏弱。作者没有提供患者心肌样本或预后队列分析,因此目前的临床相关性主要建立在其对心梗后交感-炎症-重构耦合的解释力上,而不是直接来自患者数据。不过,机制验证部分做得很扎实:文章整合了126734个单核转录组数据和7476个空间转录组位点,把远端区、边缘区和梗死区在空间上分开解析。进一步看,在TRPV1迷走感觉神经元消融后,心肌细胞差异表达基因在远端区有195个,在边缘区有333个,这两个数字对应的是区域特异性转录重编程的强度。更关键的是,作者分别从上游、中枢和下游做了三类干预:消融TRPV1迷走感觉神经元、抑制PVN的AT1aR神经元、阻断SCG内IL-1β,三者都能改善射血分数、减少瘢痕扩展并降低TH阳性交感纤维;反过来,在健康小鼠中直接向SCG注入IL-1β,又足以拉低射血分数并增大左室收缩期内径和容积。
从概念上看,这项工作至少改写了两个理解维度。其一,它把心梗后的炎症从单纯的局部坏死反应,推进为一个由外周感觉输入、中枢整合和交感节后神经共同塑造的跨器官放大过程。其二,它把边缘区重新定义为一个高度可塑的病理生态位:真正决定后续走向的,也许不只是中心坏死灶,而是这个持续接受神经和炎症双重塑形的borderzone。治疗上,这意味着至少有三类策略值得继续追踪,包括靶向TRPV1相关感觉输入、调节PVN内AT1aR神经元活性,以及局部阻断SCG内IL-1β介导的神经炎症。但边界也要讲清,这些干预目前仍停留在小鼠机制研究层面,离可转化的给药方式、剂量安全窗和人群筛选标准还有明显距离。
这项研究把心梗后的迷走感觉输入—下丘脑整合—交感神经节炎症这条隐性的心脑免疫环路从背景推到前台,并提示SCG内IL-1β及其上游神经通路可能成为抗重构的新切入口。

