CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
本文解读的研究发表在Science,题为“Divergent FOXA1 mutations drive prostate tumorigenesis and therapy-resistant cellular plasticity”。该工作围绕一个临床上的核心矛盾展开:AR轴治疗常见初期有效,但多数患者最终进入去势抵抗。作者没有把FOXA1突变当作同一种事件处理,而是按突变类别拆开验证,并用一套很标准但执行得很彻底的路线把因果链条闭合:同源小鼠与同源类器官将“突变类别”转化为可控变量;单细胞转录组与单细胞多组学定位细胞状态变化发生在哪些群体;ChIP-seq与motif解析增强子语法与因子占位;最后用遗传与药理干预把关键依赖钉死。读完你应该能抓住两件事:Class1更偏向驱动肿瘤发生,Class2更偏向诱导去势相关的细胞可塑性并推动耐药进展。
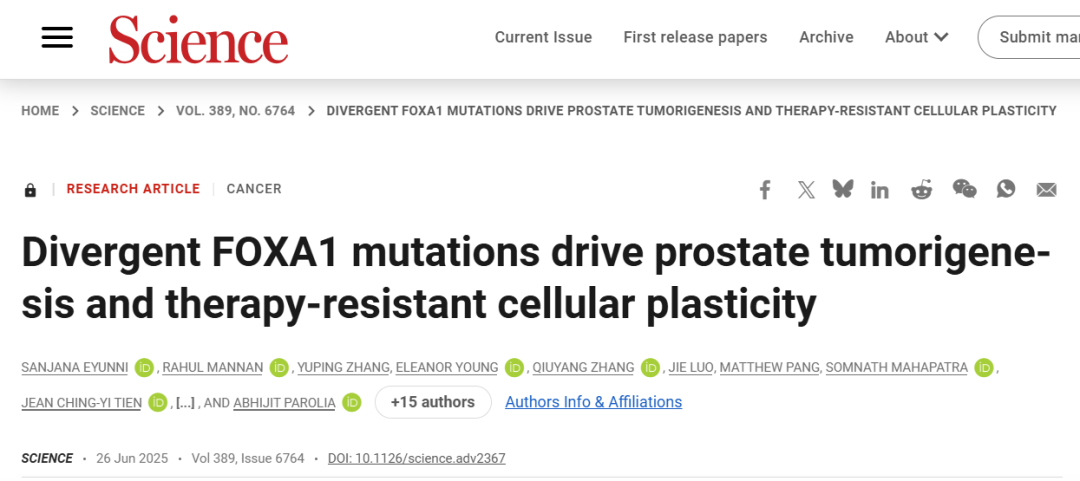
研究将FOXA1异常大体分为三类:Class1多为forkheadDNA结合域附近(wing2区域)的错义或框内缺失,既可见于原发也可见于转移;Class2多为C端移码截短,倾向在转移/去势抵抗阶段获得;Class3为基因座结构变异(如重复、易位)导致的增强子层面改变,覆盖原发与转移。
研究策略的关键在于“同平台验证”:构建可在前列腺上皮特异表达的人源突变模型,并配套同基因背景类器官;在组织学与分子层面同步使用单细胞RNA测序与单细胞Multiome(RNA+ATAC);再用ChIP-seq解析转录因子与AR的染色质占位变化;最后用去势、药物抑制与CRISPR敲除把相关性推到因果层级。
01 Class1突变在p53缺失背景下驱动高级别侵袭性腺癌形成
研究首先在体内建立Class1代表突变的条件性表达模型,使突变FOXA1在前列腺上皮细胞中被激活表达,并通过表位标记确认其定位。随年龄增长,模型动物出现从上皮增生到上皮内瘤变,再到局灶性导管内癌灶的病理演进。进一步结合Trp53缺失背景后,病变等级与侵袭性显著增强,提示Class1突变在抑癌基因缺失的背景下更容易跨越肿瘤发生门槛。
为了把“表型”落实到“细胞状态”,研究对前列腺组织进行单细胞RNA测序,在病例上皮细胞中观察到增殖相关细胞比例上升,并在转录层面呈现Ar与Foxa1相关程序增强、分化标志下调与原癌基因相关表达上移。拟时序分析进一步支持由正常腔面细胞向肿瘤样状态的连续过渡,从而把Class1突变与肿瘤发生的细胞程序改变建立了同一链条上的证据。
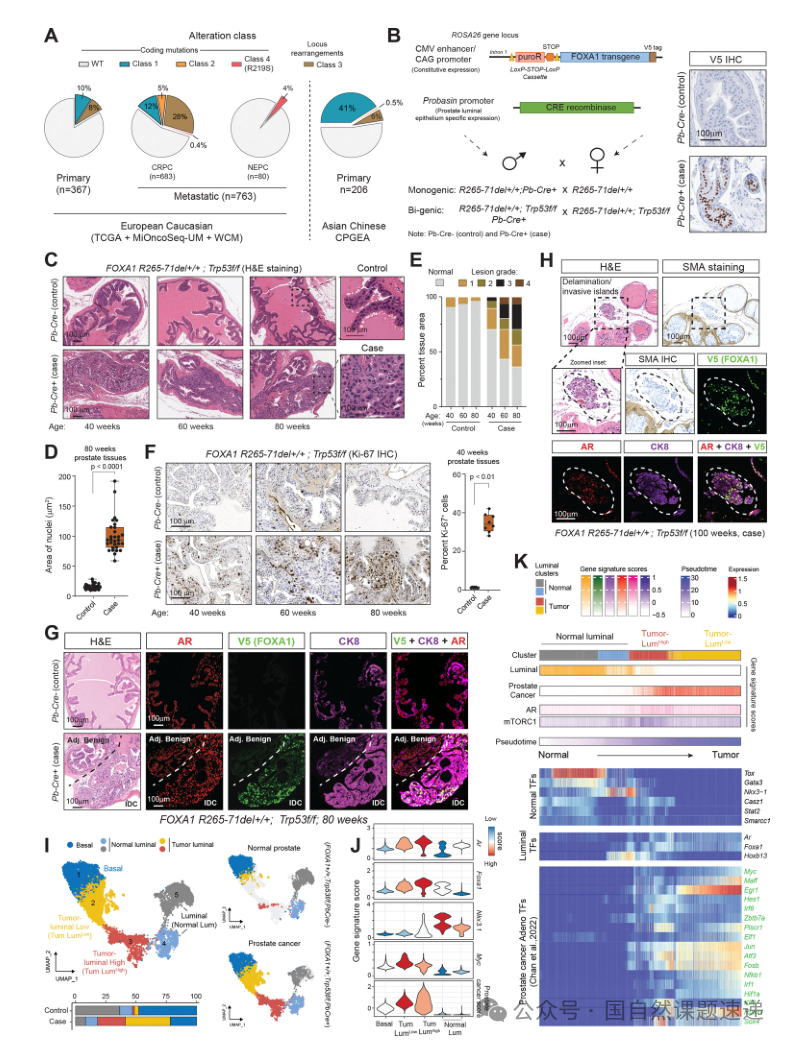
图1 FOXA1Class1突变在p53缺失背景下驱动高级别侵袭性腺癌
02 Class1突变并行激活AR与mTORC1相关致癌信号,并重写AR增强子占位图谱
研究将体内发现转入可操控的类器官体系,比较同基因背景下Cre−与Cre+类器官的信号与转录差异。结果显示,Class1突变不仅增强AR相关基因特征,也显著提高mTORC1相关通路活性,并呈现对经典AKT依赖路径不敏感的特征:mTORC1/2抑制可有效压制下游信号,而AKT抑制效应有限;同时,AR降解并不能完全消除mTOR轴的激活,提示两条通路具有并行或部分独立的致癌贡献。
在染色质层面,研究使用AR ChIP-seq发现突变背景下出现大量新增AR结合位点,且这些位点更偏向远端顺式调控区域,并富集FOXA1相关复合基序而非典型ARE回文序列,说明Class1突变通过“增强子级别重布线”改变AR的调控语法。
更重要的是,研究通过蛋白互作与功能筛选锁定NSD2作为关键协同因子:在突变类器官中抑制或敲除NSD2可显著削弱生长并抑制AR信号,同时造成AR在绝大多数位点的装载受阻,从而把“增强子重布线”从现象推进到可干预的因果机制。体内去势实验进一步表明,该类肿瘤表型对雄激素环境高度依赖,符合“AR依赖的促癌路径”。
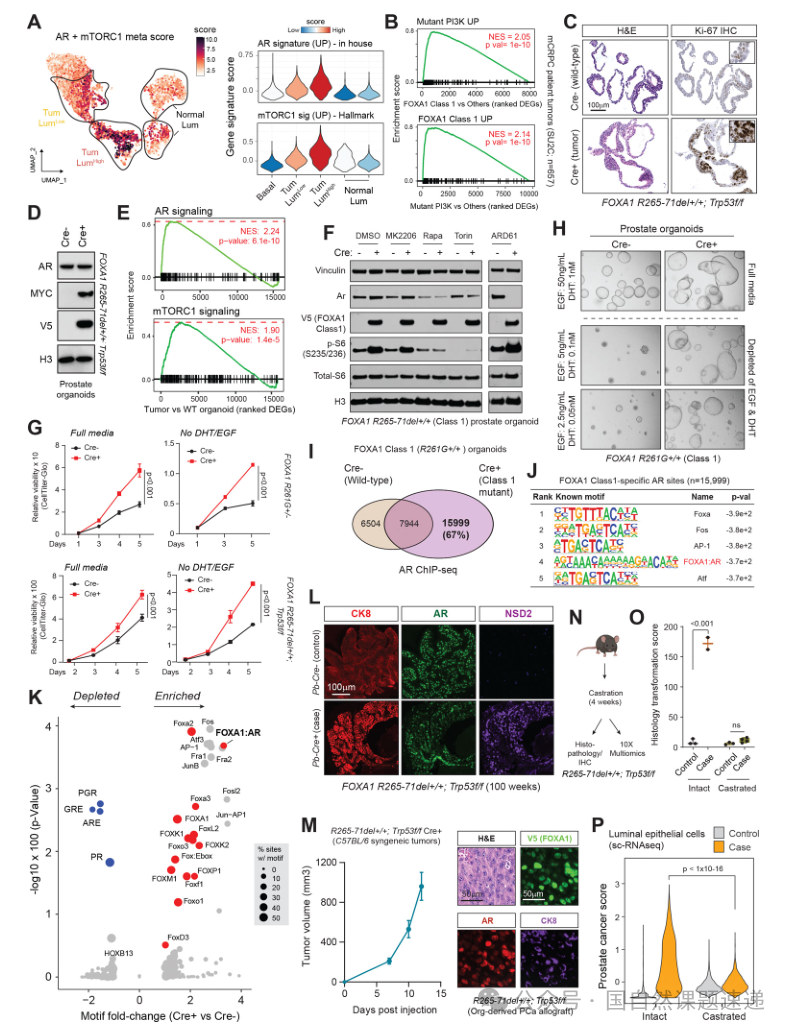
图2 FOXA1Class1突变协同激活AR与mTORC1致癌信号
03 Class2突变触发与去势相关的腔内可塑性,诱导分化腔面细胞获得干性样程序
与Class1不同,Class2代表性移码截短突变在体内并不直接诱发典型癌前病变或明显肿瘤形成,提示其主要作用不在“启动肿瘤发生”。研究转而采用单细胞Multiome对同一细胞的表达与染色质开放状态进行配对解析,发现病例上皮细胞中出现两类显著富集的病例特异细胞群:它们保留分泌型腔面细胞标志,同时上调与腔面近端祖细胞/干性相关的共识签名,被定义为由Class2突变诱导的腔面干性样细胞群。
为避免“原本就存在的祖细胞扩增”这一替代解释,研究进一步在组织切片层面用标志物免疫染色进行共定位验证,显示干性相关标志阳性细胞显著增加,并与突变FOXA1表达细胞高度重叠;同时这些细胞仍表达AR及腔面分化相关标志,支持“分化腔面细胞被重编程为具备部分祖细胞特征的状态”,即腔内可塑性增强。
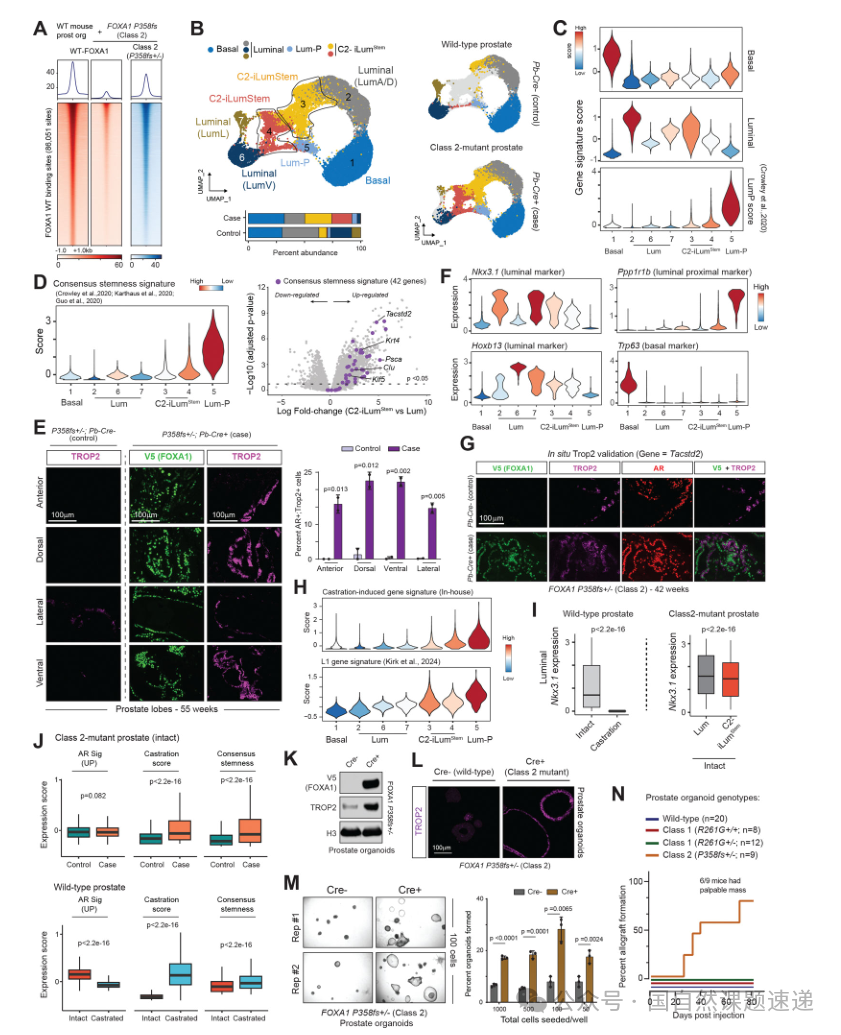
图3 FOXA1Class2突变触发与去势相关的腔内可塑性
04 Class2突变通过KLF5/AP-1新生增强子回路建立耐药表型,并赋予去势环境下存活优势
研究利用单细胞ATAC与基序富集分析显示,Class2突变相关细胞群获得大量新增开放的远端顺式元件,这些元件富集AP-1与KLF家族基序,而AR及其他核受体基序并不占优势;ChromVAR进一步提示AP-1复合体活性增强,并伴随KLF5等因子在病例细胞中的表达与活性上升。这一结果指向一条与经典AR增强子语法不同的调控体系:以AP-1与KLF5为核心驱动的“新生增强子网络”。
在机制落地方面,研究通过FOXA 1与KLF5的ChIP-seq比较野生型与Class2突变体的占位差异,发现突变体获得大量新结合位点,并与KLF5在新增位点上共占位,同时突变体还直接促进 AP-1 家族因子相关基因座的转录激活,形成自增强回路。功能层面,Class2 突变类器官在去雄条件以及AR拮抗剂处理下仍维持更强的生长与存活能力,符合治疗耐受表型。体内去势后,突变表达细胞在腔面细胞群中的比例显著上升并伴随增殖信号增强,提示治疗压力造成对耐受细胞的选择性富集,这是“耐药演进”在组织层面的直接证据。
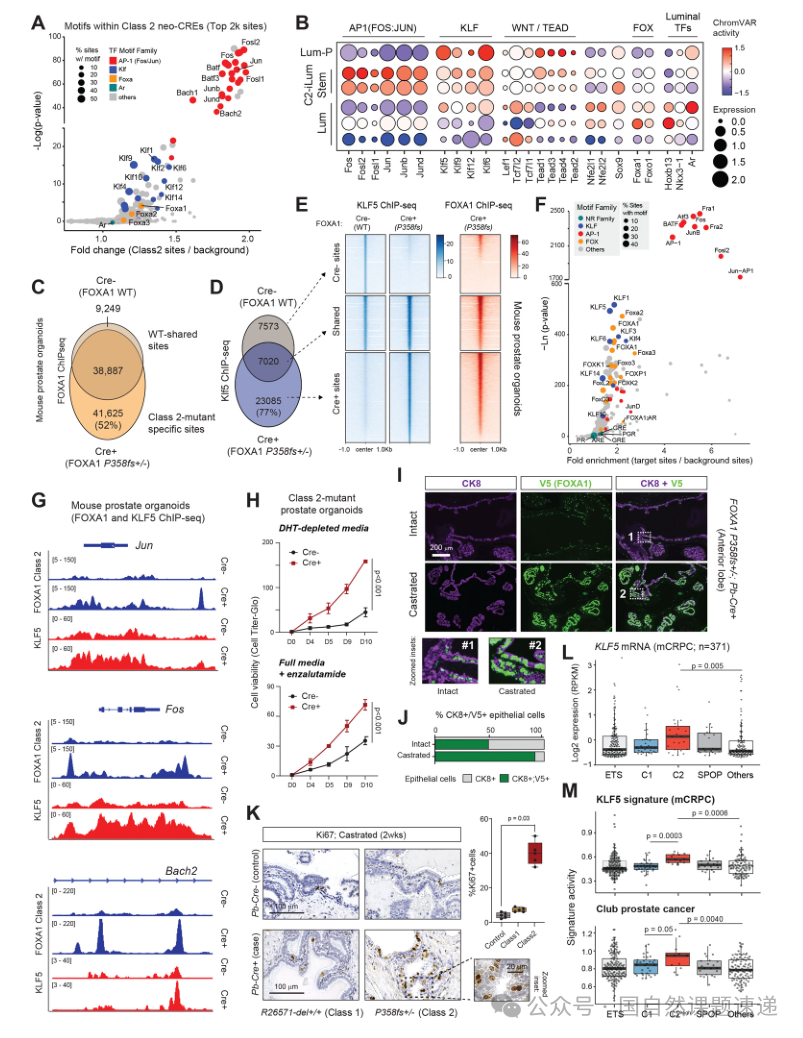
图4 FOXA1Class2突变激活KLF5/AP-1新生增强子回路驱动治疗耐受
综合证据显示,Class1突变更倾向在p53缺失等促肿瘤背景下推动AR依赖型腺癌形成,其关键特征是AR增强子占位图谱被重写,并伴随mTORC1/2等致癌信号共激活,且对NSD2等协同因子具有依赖性。相对地,Class2突变不以直接致癌为主,而是通过在分化腔面细胞中诱导腔内可塑性与干性样程序,并建立由KLF5/AP-1主导的新生增强子回路,使细胞在去势或AR通路抑制下仍能维持存活与扩增,从而为治疗后进展提供细胞与分子基础。
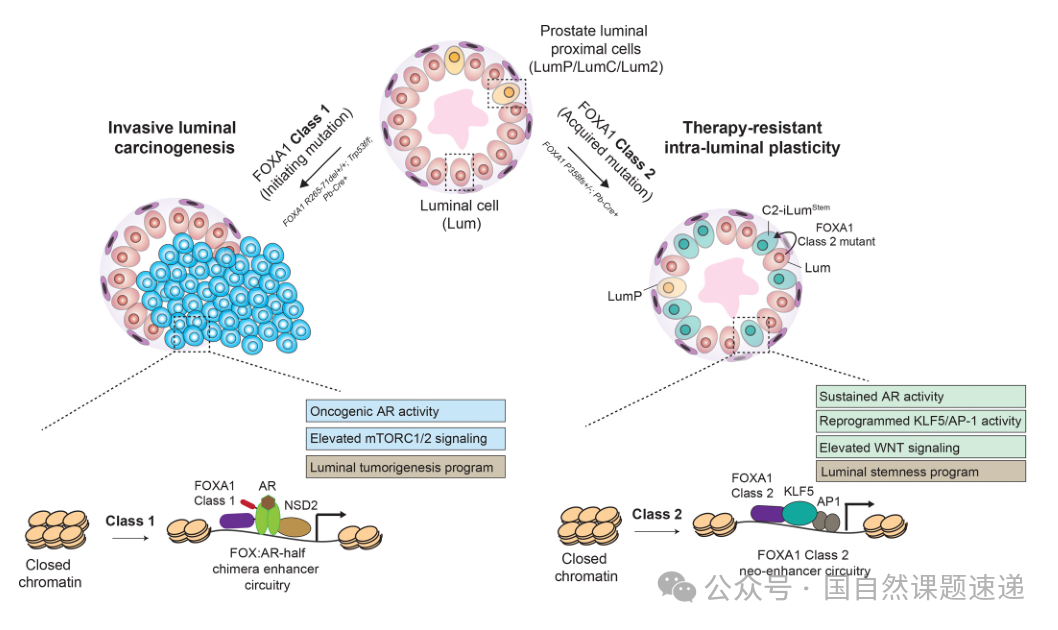
图5 FOXA1Class1与Class2突变在促肿瘤发生与治疗耐受中的不同致癌机制示意

