CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
01
研究背景与切入
肿瘤微环境与慢性抗原刺激可促使CD8⁺T细胞进入耗竭状态,表现为细胞毒性与细胞因子产生能力下降,抑制性受体持续上调,干性维持受损,从而限制免疫治疗获益。既往研究多从转录调控与表面表型出发描述耗竭谱系,但耗竭的稳定维持往往涉及更深层的细胞内稳态网络。本研究将问题下沉至蛋白质稳态层面,聚焦耗竭T细胞在长期刺激背景下如何处理持续增加的蛋白合成负荷与折叠压力,并据此寻找可干预的关键节点。
作者提出并验证耗竭T细胞存在一种特异的蛋白毒性应激反应,命名为Tex-PSR。该反应的核心特征包括全局翻译增强、分子伴侣网络上调、错误折叠蛋白与蛋白聚集体累积、应激颗粒增加,以及以自噬为主的蛋白降解与周转增强。该框架提示,耗竭并非仅由表面受体与转录程序被动表征,蛋白稳态失衡可能构成推动耗竭形成与维持的重要驱动力。
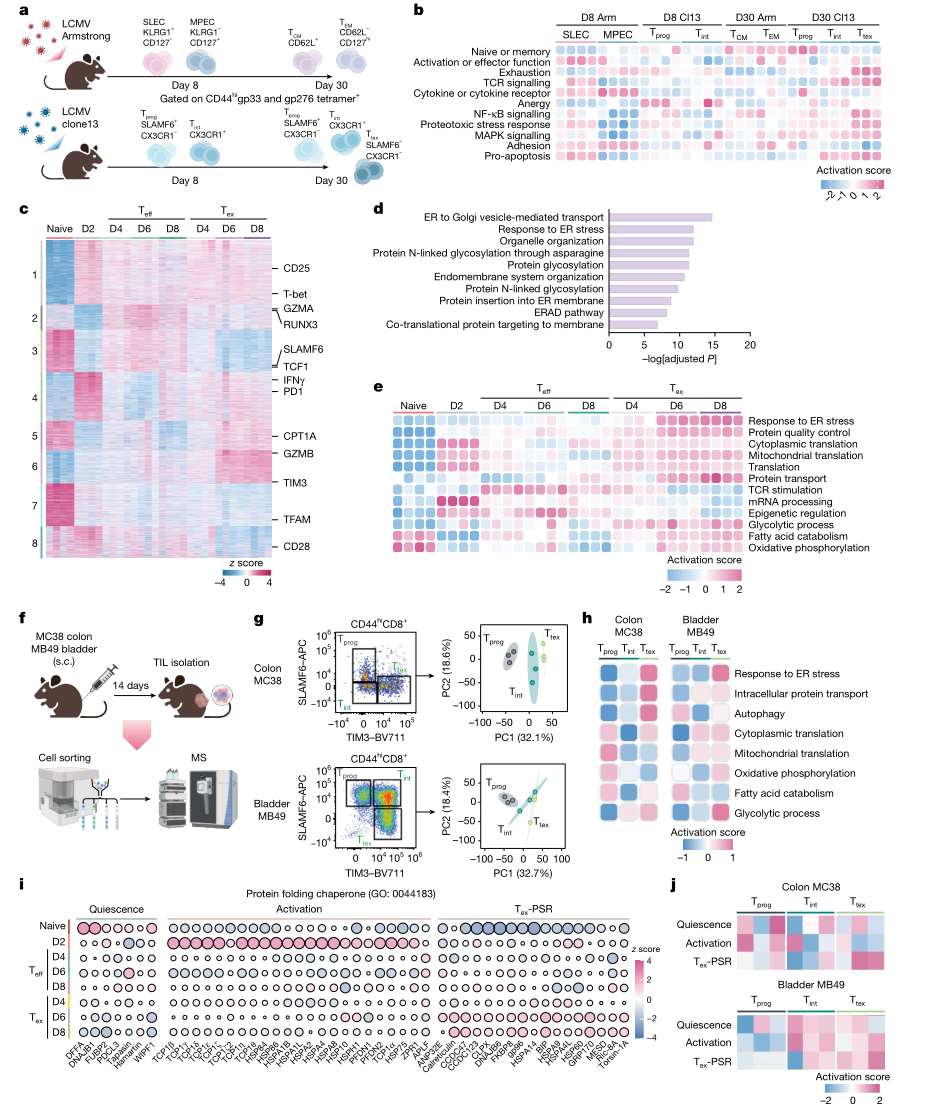
图1 蛋白毒性应激反应(PSR)在耗竭T细胞(Tex)中被触发并呈现分子伴侣蛋白的动态表达
02
研究设计与技术路线
本研究的方法学特点在于以定量蛋白质组学为主线,在多模型与多阶段的耗竭体系中进行系统对照,并通过多类正交实验将通路推断落实为可直接测量的细胞学读数。研究首先建立体外耗竭模型,通过重复TCR刺激诱导Tex,并行获取bulkRNA-seq与质谱定量蛋白质组学数据,同时结合流式细胞术与功能检测追踪耗竭表型与效应能力变化。蛋白质组学采集采用以DIA为核心的流程,并借助高覆盖的chromatogramlibrary提高鉴定深度与跨批次可重复性,以保障不同状态间差异比较的稳健性。
为验证结论的普适性,研究进一步扩展至体内慢性感染模型与肿瘤模型。慢性感染采用LCMV急性感染与慢性感染体系实现不同耗竭轨迹的对照;肿瘤体系采用MC38与MB49模型,从肿瘤浸润CD8⁺T细胞中分选不同耗竭阶段的亚群,并进行蛋白质组学定量与通路层面比较。通过上述设计,研究建立了耗竭进程分层与蛋白稳态网络重编程之间的对应关系,并为后续因果检验提供了结构化证据基础。
03
蛋白层证据:转录信息不足以替代蛋白稳态机制
研究首先在数据层面显示,Teff与Tex中mRNA与蛋白表达一致性整体有限,且不同通路的脱耦程度存在显著差异。免疫信号与部分效应相关模块在转录与蛋白层面相对一致,而代谢、转录后调控、表观调控与蛋白稳态相关模块更易出现转录与蛋白表达不同步。该结果强调,耗竭过程中与翻译、折叠、伴侣体系与降解通路相关的关键变化可能主要发生在蛋白层面,仅依赖转录组难以完整捕捉Tex-PSR所代表的蛋白稳态重塑过程。
04
Tex-PSR表型刻画:从聚集、翻译到周转的多维验证
研究围绕蛋白毒性负荷的形成与处理,构建了由聚集体、翻译活性与蛋白周转组成的多层验证体系。首先在蛋白聚集体层面,作者利用可结合错误折叠蛋白富集结构的探针检测,在体外Tex以及体内肿瘤浸润CD8⁺T细胞中均观察到聚集体信号升高,并随耗竭加深呈递增趋势,提示蛋白错误折叠与聚集在耗竭进程中持续累积。
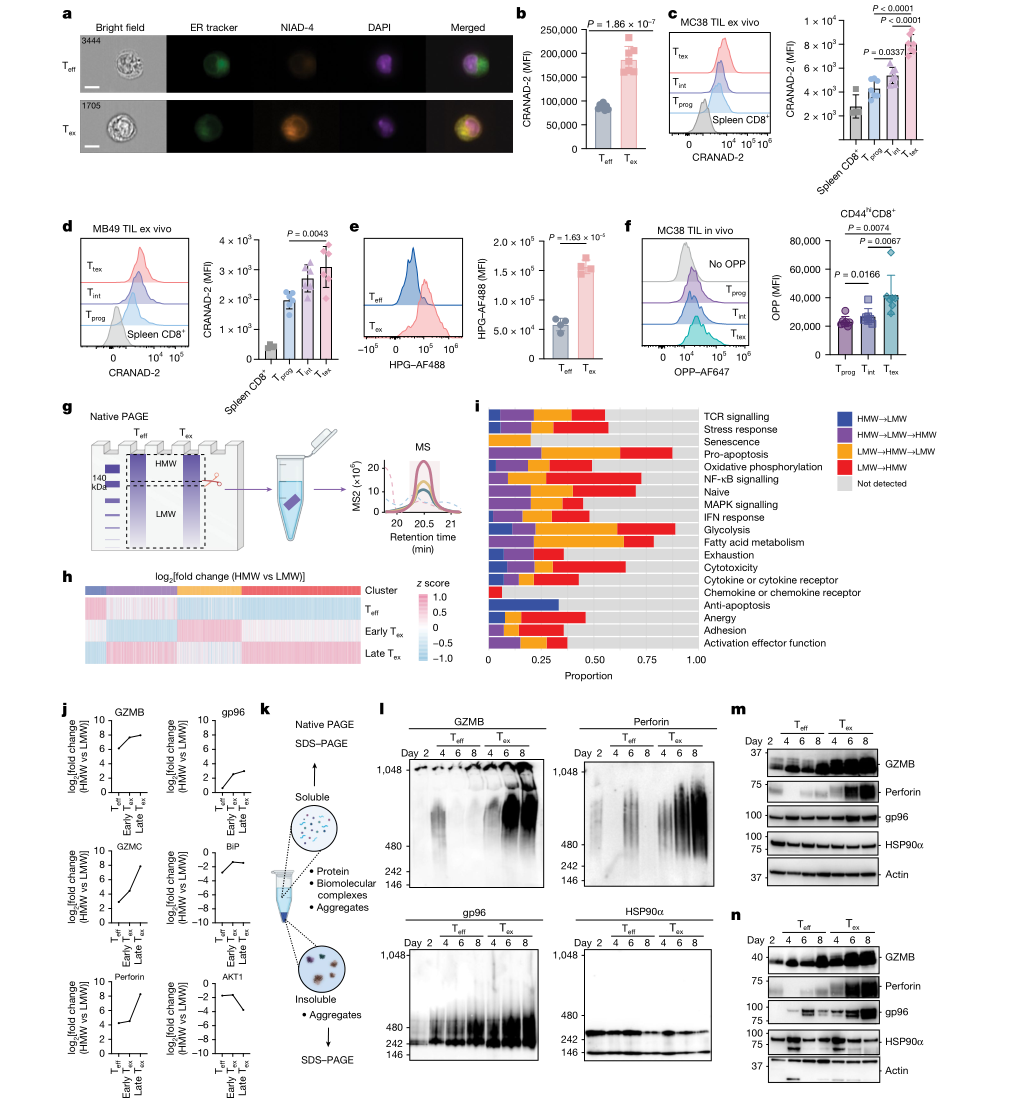
图2 慢性TCR刺激在T细胞耗竭过程中破坏蛋白质稳态
其次在翻译活性层面,研究采用新生蛋白标记策略对全局翻译进行直接测量。体外使用HPG掺入评估新生蛋白合成,体内在荷瘤模型中使用OPP标记延伸肽链,结果一致显示Tex,尤其终末耗竭群体,具有更高的总体翻译水平。该发现提示Tex-PSR并不表现为翻译的普遍抑制,而是在高翻译输出的背景下伴随更强的蛋白质质量控制需求。
进一步在蛋白周转层面,研究通过HPG脉冲追踪评估新生蛋白降解动力学,显示Tex中新生蛋白更快进入降解通路,且抑制实验支持自噬在该过程中占据主导地位。由此可见,耗竭T细胞处于高合成与高降解并存的高周转状态,通过增强清除能力缓冲持续翻译带来的蛋白毒性负荷。与此同时,研究观察到Tex中应激颗粒增加,并通过关键组分干预提示应激颗粒与细胞功能输出及生存维持之间存在可分离效应,从而进一步支持Tex-PSR是一套兼具“负荷缓冲”与“功能代价”的稳态适应机制。
05
因果关系验证:蛋白毒性可直接驱动耗竭程序
本研究的关键在于对因果关系的检验。作者在排除仅以相关性解释的基础上,采用两种互为补充的策略验证蛋白毒性压力对耗竭程序的驱动作用。其一,通过化学方式诱导错误折叠与聚集压力,即使在缺少典型慢性重复刺激的条件下,仍可促使细胞出现更明显的耗竭表型与功能受损,并在体内转移体系中呈现更偏终末耗竭的分化趋势。其二,在T细胞中表达易聚集且功能上相对惰性的突变蛋白CFTR(ΔF508),同样可在非慢性刺激背景下诱导耗竭相关特征,并在人源CD8⁺T细胞中推动耗竭相关群体比例上升。两条策略从不同角度共同支持,蛋白稳态破坏本身可构成耗竭形成的重要驱动因素。
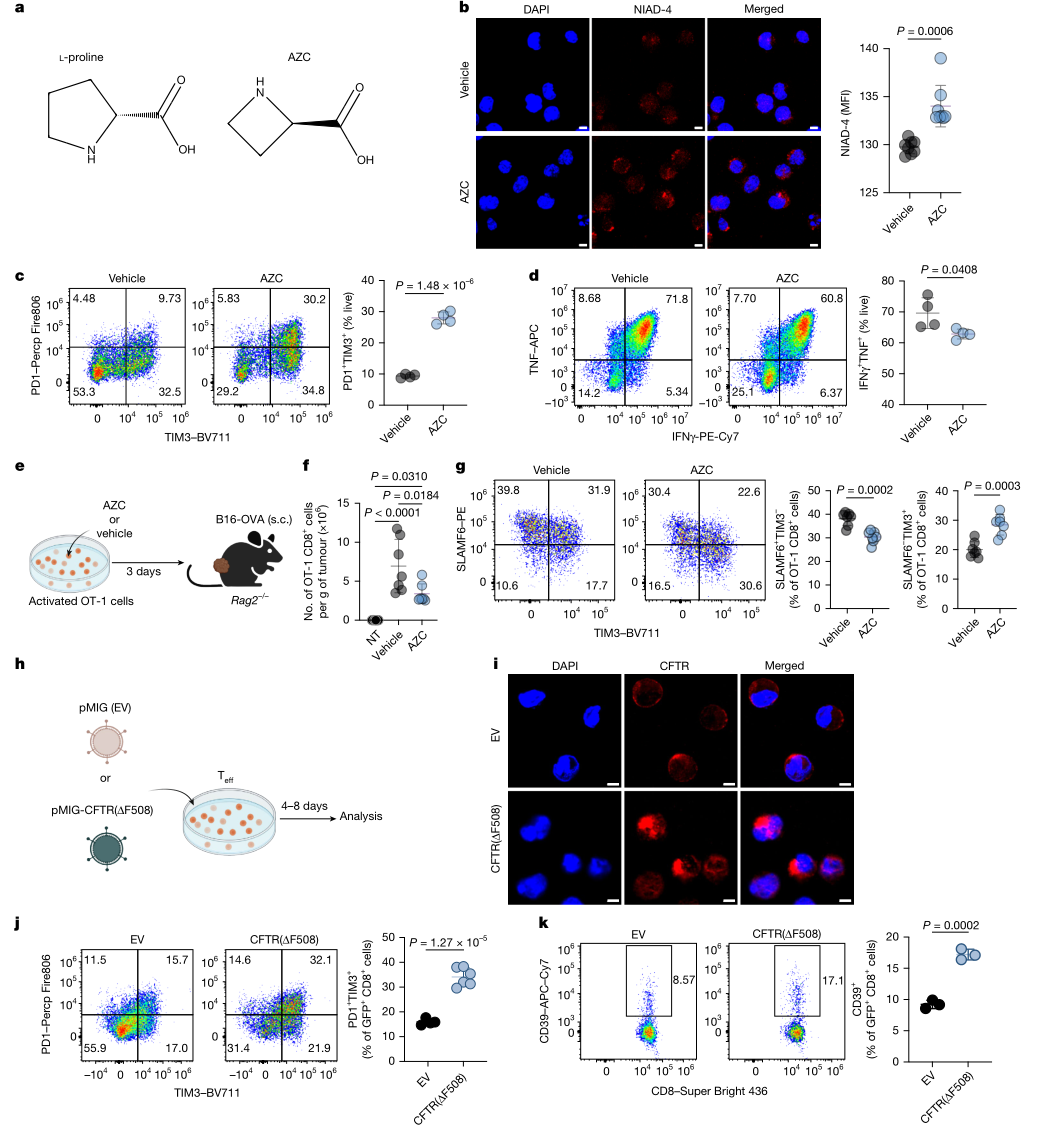
图3 蛋白错误折叠在无慢性TCR刺激条件下也可促进T细胞耗竭
06
上游信号枢纽:持续AKT连接翻译负荷与Tex-PSR
在明确Tex-PSR的存在及其因果贡献后,研究进一步追踪其上游调控。蛋白质组学与信号读数提示AKT轴在晚期耗竭状态中更为活跃。药理学干预显示,适度AKT抑制能够在不显著损害增殖的条件下改善干性相关表型,恢复细胞因子产生,并同步降低翻译速率、聚集体水平与伴侣蛋白表达。与此相对,构成型活化的myrAKT可在非耗竭条件下诱导Tex-PSR相关表型,推动细胞向终末耗竭方向偏移,并在肿瘤模型中表现为浸润减少与控瘤能力下降。上述结果提示,持续AKT信号可将高翻译输出与蛋白稳态压力长期耦合,从而在机制层面支撑耗竭的形成与维持。
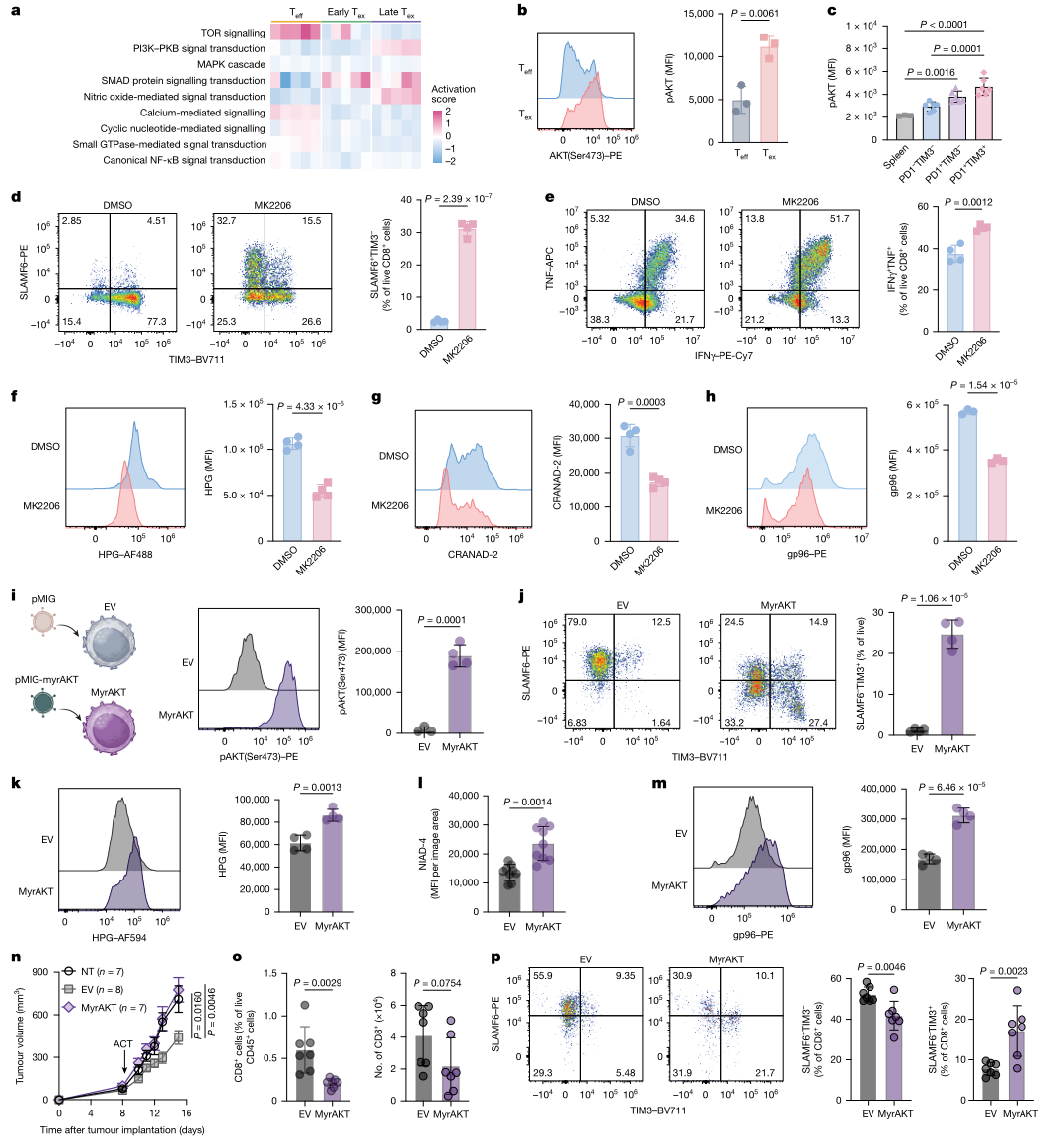
图4 持续AKT信号诱导Tex-PSR并构成T细胞耗竭的关键基础
07
靶向验证:抑制Tex-PSR关键伴侣节点可改善抗肿瘤效应
围绕可转化干预,研究将靶点从上游信号进一步下沉至Tex-PSR执行层的蛋白折叠与伴侣网络。作者采用CRISPR对候选蛋白稳态相关基因进行功能筛选,在重复刺激诱导体系中评估细胞因子输出与耗竭表型改变。结果显示,对Hspa5(BiP)、Hsp90b1(gp96)与Ero1a等关键节点的干预可显著提升细胞因子产生,并推动表型向更具干性与效应潜力的方向移动。体内验证进一步表明,删除Hspa5或Ero1a的抗原特异T细胞可在肿瘤模型中带来更优的控瘤表现,并呈现更有利的细胞毒性相关群体特征与干性维持信号。该部分工作将蛋白稳态机制与疗效改善建立直接连接,为后续治疗策略提供实证基础。
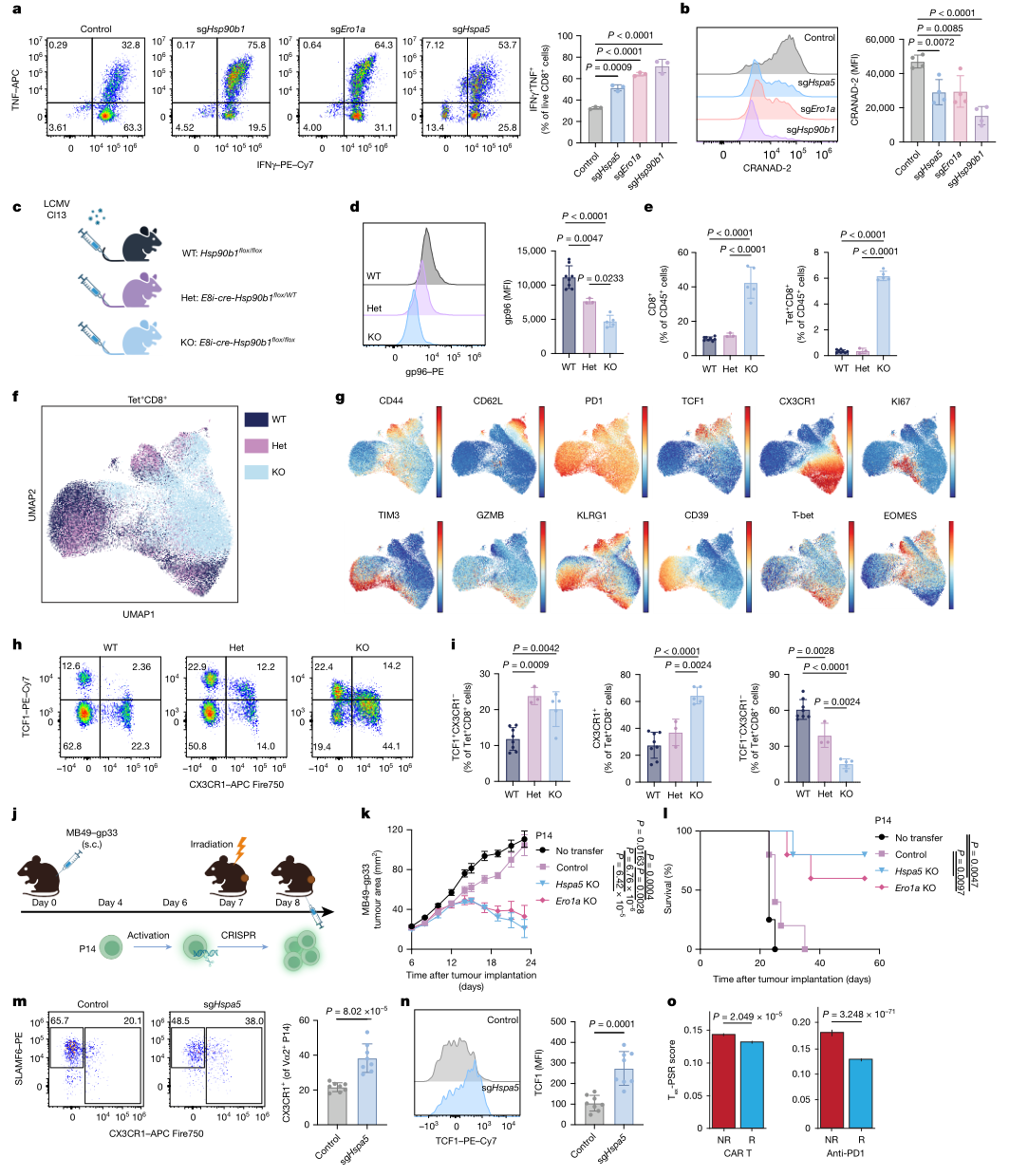
图5 靶向Tex-PSR分子伴侣可抑制T细胞耗竭并增强抗肿瘤免疫治疗
08
人群数据关联:Tex-PSR具有分层与预测价值
研究进一步利用公开的人类pan-cancer单细胞数据构建Tex-PSRsignature,并在细胞亚群与分化轨迹层面评估其动态变化。结果显示,Tex-PSR特征在终末耗竭群体中最为显著,并在耗竭分化早期出现且随耗竭加深增强。进一步在临床队列与治疗队列的关联分析中,较高的Tex-PSR特征与较差的总体生存趋势以及较弱的免疫治疗反应相关,覆盖CAR-T与ICB等不同治疗场景。该结果提示Tex-PSR不仅是实验模型中的机制现象,亦具有潜在的患者分层与疗效预测意义。
思维导图
结论与展望
本研究以蛋白质组学为核心证据链,在体外与体内多模型、多阶段体系中揭示耗竭T细胞存在特异的蛋白毒性应激反应Tex-PSR,并通过化学诱导与聚集蛋白表达等策略完成因果验证,进一步将持续AKT信号确立为连接翻译负荷与Tex-PSR的上游枢纽,并证明靶向关键分子伴侣节点可改善体内抗肿瘤效应。总体而言,该工作将耗竭的持续驱动从传统的转录与表面表型叙事推进至蛋白稳态失衡的机制层,为免疫治疗联合策略提供了新的可干预轴线。面向转化应用,蛋白稳态网络在多组织中具有高度保守性,系统性干预的安全性窗口与递送特异性仍需严格评估。更具可行性的方向可能集中于细胞治疗制备阶段的工程化干预与短程调控,结合Tex-PSRsignature实现患者分层、疗效监测与联合方案优化,以进一步提升免疫治疗的稳定获益。
参考文献:Wang Y, Ma A, Song NJ, Shannon AE, Amankwah YS, Chen X, Wu W, Wang Z, Saadey AA, Yousif A, Ghosh G, Mandula JK, Velegraki M, Xiao T, Wen H, Huang SC, Wang R, Beusch CM, Dawood AS, Gordon DE, Abdel-Hakeem MS, Ghoneim HE, Xin G, Searle BC, Li Z. Proteotoxic stress response drives T cell exhaustion and immune evasion. Nature. 2025 Nov;647(8091):1025-1035. doi: 10.1038/s41586-025-09539-1. Epub 2025 Oct 1. PMID: 41034580; PMCID: PMC12657239.

