CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

阿尔茨海默病、帕金森病、亨廷顿病并不是真正独立的三种病,而是同一套代谢系统逐级崩溃的三种表现形式。文章提出了一个新的视角:以线粒体为中心,把内质网、溶酶体、脂滴等细胞器和葡脂代谢拉进同一个网络,用MMH网络来重写我们对神经退行性疾病的理解。
从“病理蛋白”挪到“代谢柔性”
传统课本里,神经退行性疾病总是被拆成不同模块:AD看Aβ和P-tau,PD看α-突触核蛋白和黑质多巴胺神经元,HD看mHTT突变。但这篇文章直接换了一个问题:大脑的能量系统,到底是在哪个环节开始撑不住的。作者从线粒体结构损伤、ATP合成下降、ROS堆积、钙稳态失衡讲起,一路追到葡萄糖利用率下降、脂类代谢紊乱、胰岛素抵抗和慢性低度炎症,最后把这些环节收束到一个统一框架:MMH网络,也就是“以线粒体为核心的多细胞器-能量代谢-葡脂稳态网络”。在这个框架下,所谓“不同疾病”,更多像是同一套网络在不同节点崩溃后的三种结局。
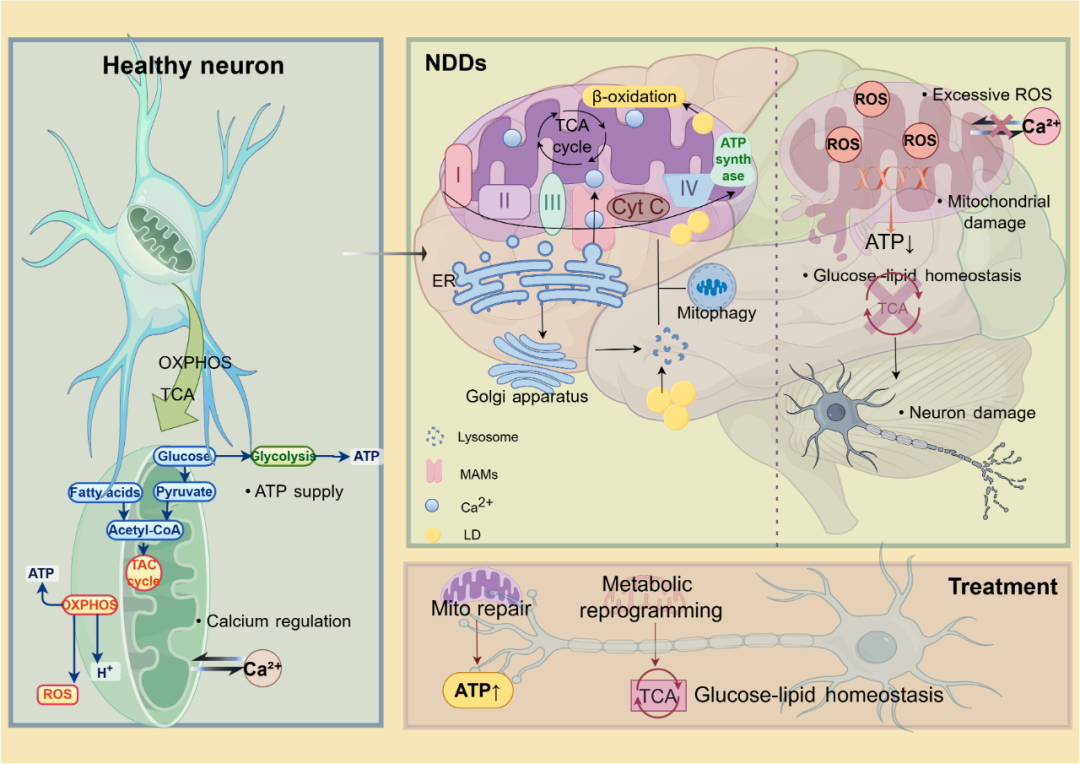
图1 以线粒体为中心的多细胞器–能量代谢–葡脂稳态调控网络。
健康神经元通过正常OXPHOS和TCA循环维持ATP供应、钙稳态和葡脂平衡;线粒体功能受损后,ROS过量、ATP下降和钙失衡拉开代谢紊乱的序幕,并与葡脂失衡形成恶性循环,最终推动神经元退行;同时内质网、高尔基体、溶酶体等通过MAMs或脂滴进一步放大线粒体代谢障碍,MMH网络理论据此提出“修复线粒体+重构葡脂代谢”的系统治疗思路。
线粒体到底在崩什么
文章第二部分回到线粒体本体,把结构、动力学和质量控制三件事捋清楚。神经元里线粒体体积分数很高,双膜结构外加大量嵴,决定了它必须保持极高的结构完整性才能撑起高耗能状态。一旦嵴塌陷、膜电位下降、复合物装配出问题,ATP就会立刻“断供”,同时ROS会急剧上升。更麻烦的是,神经元轴突又长,线粒体需要在树突和轴突之间不断运输,这就逼着融合和分裂这套动力学机制始终处在高负荷运转。MFN1/2和OPA1负责融合,DRP1、FIS1、MFF负责分裂,本来是一个“边修边开车”的自稳系统,但在NDD里这一平衡明显偏向碎片化,网络完整性被打断,损伤线粒体又难以及时被清除。
质量控制层面,PINK1/Parkin介导的线粒体自噬本来是专门处理“坏机器”的回收线,膜电位一掉,PINK1就会积累在外膜,招募Parkin打上泛素标签,再交给自噬系统打包回收。问题在于AD、PD这些疾病里,PINK1/Parkin这条线本身就被各种病理蛋白堵塞,结果就是坏掉的线粒体越堆越多,ROS和炎症因子源源不断释放,整个神经元被拖进长期应激状态。
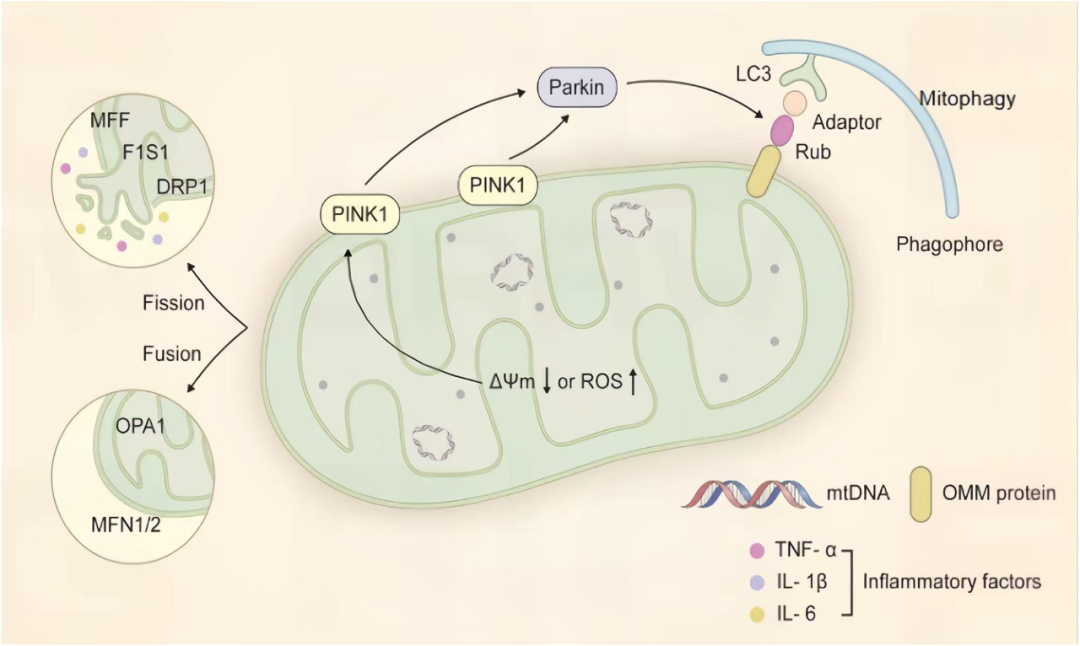
图2 线粒体结构与质量控制示意图。
图中展示了线粒体双膜结构、嵴和基质,左侧小圆示意MFN1/2与OPA1调控的融合以及DRP1、FIS1、MFF介导的分裂平衡;膜电位下降或ROS上升时,PINK1在损伤线粒体表面积累并招募Parkin,通过泛素化触发线粒体自噬;右下角标出了TNF-α、IL-1β和IL-6等炎症因子释放,提示线粒体功能失衡与NDD中炎症反应之间的病理联系。
葡脂代谢失控,炎症接管大脑
接下来文章把视角从“线粒体自己坏了”推到“整个代谢网络怎么跟着一起崩”。一方面,胰岛素信号在中枢变钝,GLUT4和GLUT3转位减少,糖酵解关键酶活性下降,丙酮酸越来越难进TCA循环,被迫转成乳酸,局部酸化、中间代谢物紊乱,神经元长期处在低能状态。另一方面,高糖环境下AGEs不断积累,和RAGE结合后直接点燃NF-kB、p38 MAPK、JNK等炎症通路,既放大细胞因子风暴,又进一步压垮线粒体,形成“糖代谢差→AGEs多→炎症强→线粒体更差”的闭环。
脂代谢这边也没有好到哪里去。β氧化效率下降,长链酰基肉碱、神经酰胺等中间体堆积,既毒神经元,又继续干扰胰岛素信号。膜脂成分的改变会直接影响离子通道、受体簇和脂筏结构,突触传递变得越来越不稳定;胆固醇稳态被打乱以后,β分泌酶被促进、α分泌酶被压制,Aβ的生成和清除彻底失衡。
小胶质细胞和星形胶质细胞在这个过程中完成了一次“代谢-表型联动”:原本依赖OXPHOS、偏修复的M2样小胶质细胞被推向高糖酵解、强炎症的M1态;星形胶质细胞的乳酸供给链断裂,从“能量队友”转变为“炎症放大器”。代谢压力+炎症信号叠加以后,大脑进入一种慢性低度炎症状态,作者用“metaflammation”来形容这种既不像急性感染、又从不真正结束的炎症背景。

图3 葡脂代谢失衡与神经炎症的交互机制。
图中左上角是代谢失调起点:高糖高脂环境诱导胰岛素抵抗,抑制CPT1和LCAD等关键酶,导致脂质中间体堆积并伴随AGEs-RAGE信号激活;右上角显示NLRP3炎症小体被激活后,小胶质细胞由M2向M1转化,大量释放TNF-α、IL-1β和IL-6;右下角是氧化应激和脂质过氧化,ETC功能下降和膜电位减弱推动4-HNE和MDA产生;左下角描绘线粒体通透性转换孔开放、钙超载和自噬受损,四个模块共同构成代谢紊乱-炎症-氧化应激-线粒体损伤的正反馈回路。
同一套代谢系统崩溃的三种“版本”
文章把AD、PD、HD放在同一个坐标系下做了对比。AD这条线,核心是Aβ沉积和P-tau异常磷酸化与线粒体直接对接:一方面Aβ和ABAD、复合物IV结合,拉低膜电位、促使mPTP开放,引发钙超载和细胞色素c释放;另一方面,来自损伤线粒体的ROS又反向激活GSK-3β、CDK5、JNK等激酶,继续推进tau异常磷酸化,形成“病理蛋白-线粒体-病理蛋白”的闭环。
PD则更偏向一个“复合物I靶向损伤+蛋白聚集”模型。黑质复合物I活性被下调三四成,其他复合物相对还算稳定,这导致局部ATP供应不足、ROS显著升高,再叠加PINK1/Parkin缺陷,损伤线粒体难以及时清除。心磷脂和α-突触核蛋白的相互作用被提出为新机制:心磷脂一方面维持线粒体膜稳定,另一方面又参与调节α-突触核蛋白构象和聚集倾向,给未来靶向干预留了一个切入口。
HD则是最典型的“全局崩溃”:mHTT直接贴在线粒体外膜上,增加膜通透性,促进细胞色素c外泄;与此同时它压制PGC-1α,线粒体生成受阻,并推动分裂多于融合,让线粒体网络彻底碎片化。影像学上可以看到,能量代谢下降早于临床症状,在无症状携带者身上就已经存在,说明能量系统故障在HD里是一个超前信号。
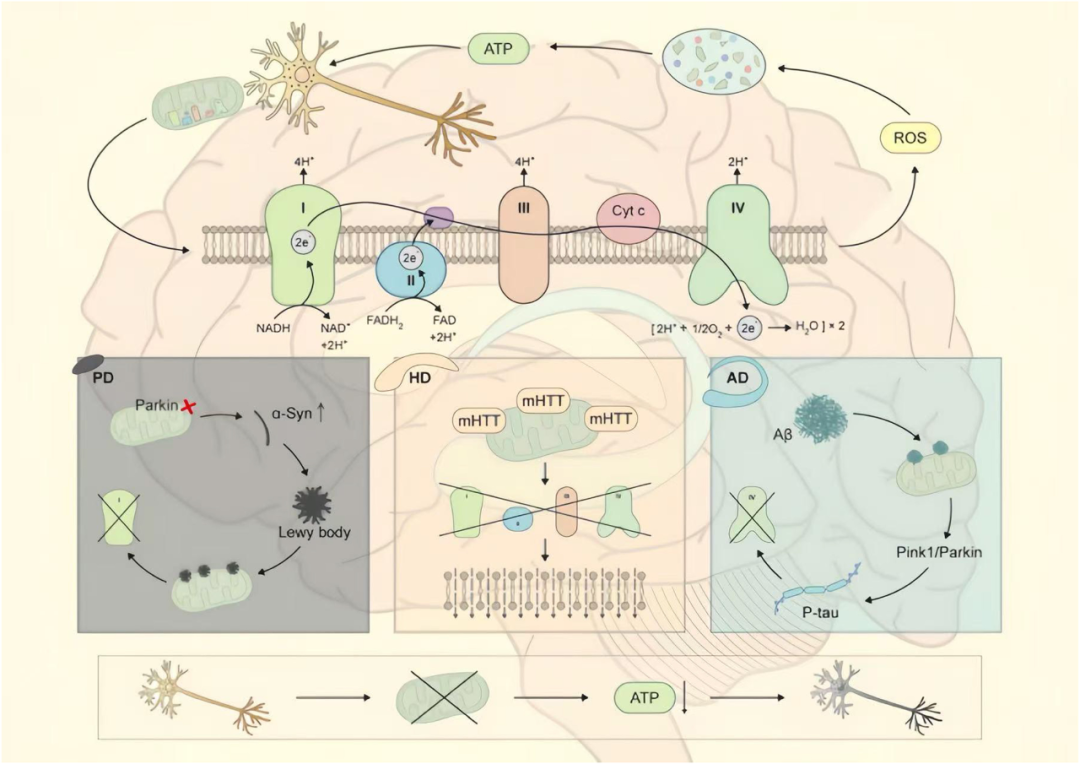
图4 三种神经退行性疾病中线粒体功能障碍的差异。
上半部分展示正常线粒体呼吸链复合物I-IV的电子传递过程;下方三个模块分别代表PD、HD和AD的特异病理机制:PD模块中Parkin功能障碍导致α-突触核蛋白聚集为路易小体,主要损伤复合物I;HD模块中mHTT破坏线粒体膜并引发整条呼吸链功能受损;AD模块中Aβ聚集与P-tau协同,优先影响复合物IV,并干扰PINK1/Parkin介导的质量控制。底部流程图总结共同结局:正常神经元→线粒体受损→ATP生成下降→神经元退行。
AMPK–mTOR–PGC-1α–SIRT1轴
作者在后半篇把重点放在三条关键信号轴:AMPK/mTOR、PGC-1α和SIRT1。AMPK是能量守门员,检测AMP/ATP比例,在“能量紧张”时启动节能模式:促进GLUT4转位、提升脂肪酸氧化、抑制脂肪酸合成,同时通过磷酸化PGC-1α拉动线粒体生物合成;mTORC1则偏向合成和生长,在能量充足时推动蛋白和脂质合成,并抑制自噬。两者互相牵制,维持一个动态平衡。
PGC-1α被称作“线粒体程序员”,和NRF1/2、PPARs等结合,同时管mtDNA复制、呼吸链复合物装配、TCA循环酶表达和抗氧化防线。SIRT1坐在这套系统上游,靠NAD+驱动去乙酰化PGC-1α、FOXO3a等靶标,一边提升线粒体生成和自噬,一边调脂、稳胆固醇。文章特别强调,随着年龄和疾病进展,NAD+下降→SIRT1活性降低→PGC-1α信号减弱→线粒体功能持续滑坡,这一条链很可能是衰老相关NDD的共性主线。
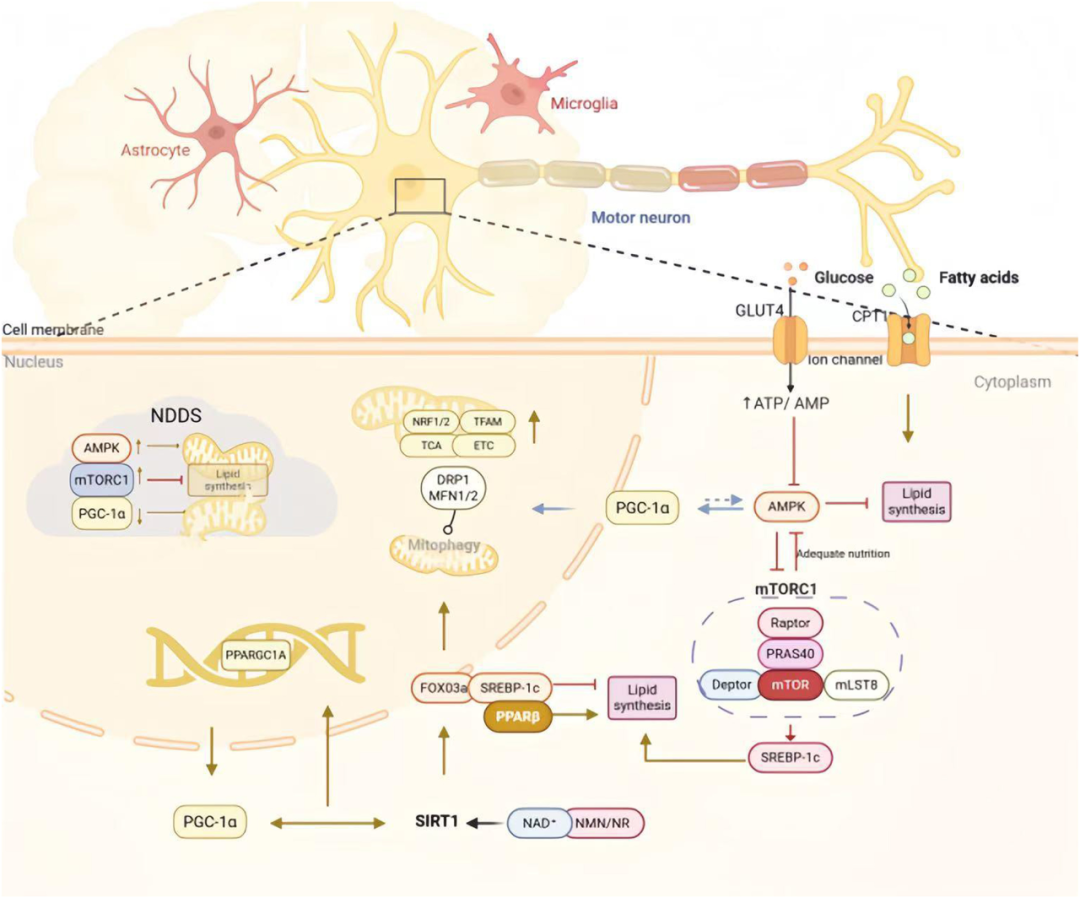
图5 AMPK/mTOR、PGC-1α与SIRT1在神经元线粒体功能和葡脂代谢中的协同调控。
图上方展示神经元及其微环境,细胞膜上的GLUT4和CPT1负责葡萄糖与脂肪酸摄取;细胞内以AMPK与mTORC1的动态平衡为核心:AMPK激活时促进GLUT4转位和脂肪酸氧化,抑制脂质合成并压制mTORC1;营养充足时mTORC1活化,通过SREBP-1c促进脂质合成,并反馈抑制AMPK。PGC-1α通过AMPK磷酸化和SIRT1去乙酰化双重激活,统筹线粒体生物合成和动力学;SIRT1可被NMN/NR激活,一方面通过FOXO3a促进线粒体自噬,一方面抑制SREBP-1c并激活PPARα等转录因子。左侧NDDS模块指出:AMPK过度激活会带来异常分裂,mTORC1过度激活会抑制自噬并促进异常蛋白聚集,PGC-1α和SIRT1下降则共同加重线粒体功能障碍。
MMH网络带来的真正转变
如果只看单个片段,这篇文章讲的内容很多人都听过:线粒体受损、胰岛素抵抗、脂质过氧化、小胶质细胞激活等等。真正有价值的地方是,作者强行把这些碎片重新拼成一个闭环:神经退行性疾病的本质,是MMH网络的代谢柔性被一步步耗尽。当糖代谢、脂代谢、炎症、线粒体、细胞器互作连成一个自我放大的正反馈,任何单一靶点药物都会显得力不从心。
对做基础研究的团队来说,这篇综述等于在提醒:选题不必永远纠结“再找一个新蛋白”,而是可以从“打断哪个反馈环、恢复哪一段代谢柔性”入手;对做转化和临床的团队,MMH网络提供了一个思路:早期用代谢-影像-体液标志物去捕捉能量失衡的信号,尽可能在结构不可逆损伤之前把系统拉回来。
最后一句话概括这篇文章:AD、PD、HD这些名字只是临床标签,底层真正出问题的,是那台负责给大脑供电、调度底物和处理垃圾的“多细胞器操作系统”。什么时候我们能真正修好这套系统,才有可能改写神经退行性疾病的自然病程。

