CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
01 原理
Western Blot(蛋白质免疫印迹法)是一种常用于科研中的蛋白质分离和鉴定技术。通过该技术,将混合的蛋白质样品依据分子量(即蛋白质类型)进行凝胶电泳分离。分离后的结果会转移到膜上,在每个蛋白质上形成对应的条带。然后,通过将特定的抗体与膜上的蛋白质反应,未结合的抗体会被洗去,只留下结合到目标蛋白质上的抗体。通过显影膜,可以检测到这些结合的抗体。
由于抗体只与目标蛋白结合,因此在膜上只会看到一条条带。条带的厚度与蛋白质的含量成正比,借助标准品可以测定目标蛋白的浓度。
02 贴壁细胞操作步骤
1、使用冷的磷酸盐缓冲盐水(PBS)清洗细胞培养瓶或培养皿,轻轻摇动以确保细胞被彻底清洗。弃去PBS。(提示:整个过程保持培养皿在冰上进行,以防止蛋白质降解。)
2、向培养皿中加入PBS,使用细胞刮刀将细胞从培养皿表面刮离。将细胞悬液转移到微量离心管中。
3、以1500RPM离心5分钟,弃去上清液。
4、加入180μL冰冷的细胞裂解缓冲液,并加入20 μL新鲜的蛋白酶抑制剂混合液。(提示:如果最终提取的蛋白浓度较低,建议使用更多比例的蛋白酶抑制剂,重复此步骤。)
5、将裂解液在冰上孵育30分钟,随后在4°C下以12,000 RPM离心10分钟,澄清裂解液。
6、将上清液(或蛋白混合液)转移到新的管中,并存放在冰上或冷冻于-20°C或-80°C。
7、使用分光光度计测量蛋白浓度,以确保后续分析所需的蛋白质量。
03 样品准备
1、使用浓度与质量的关系公式,计算所需的蛋白提取液量,以确保每个孔中有50 μg的蛋白。
2、向样品中加入5 μL样品缓冲液,并使用去离子水(dd H2O)调整体积,使每个孔的体积一致。混匀。(提示:每个孔的总液量建议为15 μL)。
3、将样品放置在干热板上加热5分钟,温度为100ºC。
04 凝胶制备
10% 堆积胶
dd H2O 3 mL
1 M Tris–HCl 2.1 mL (pH 8.9)
30% Acr Bis 2.8 mL
10% SDS 80 µL
10% APS 56 µL
TEMED 6 µL
6% 分离胶
dd H2O 2 mL
1 M Tris–HCl 400 µL (pH 6.7)
30% Acr Bis 600 µL
10% SDS 36 µL
10% APS 24 µL
TEMED 4 µL
注:APS:过硫酸铵;TEMED:四甲基乙烯二胺;SDS:十二烷基硫酸钠
1、准备好10%的堆积胶溶液后,组装凝胶固化架(图1)。
提示:10%的APS和TEMED使溶液固化,因此可以同时准备两种胶,如果上述试剂直到最后才添加,效果会更好。
2、小心地将堆积胶溶液加入,直到其液面达到玻璃板上的绿色标线(图2),然后加入水。等待15至30分钟,直到胶凝固。
提示:使用吸管可以使添加凝胶到玻璃板的过程更加简便。
3、在移除水后,将堆积胶与分离胶叠加。
提示:最好将设备倾斜,并使用纸巾去除水分。
4、插入梳子,确保没有气泡。
5、等待胶完全固化。
提示:可以通过将一些凝胶溶液留在管中检查固化是否完成。

图1. 支撑凝胶在固化过程中保持稳定的装置。

图2. 使用移液管添加凝胶溶液。
05 电泳
1、将运行缓冲液倒入电泳槽中(图3)。
2、将凝胶放入电泳槽中,并连接到电源。
提示:连接电源时,确保红色连接到红色,黑色连接到黑色。
3、确保缓冲液完全覆盖凝胶,然后小心地移除梳子。
加载分子标记(6 µL)和样品(15 µL)到每个孔中(图4)。
4、以低电压(60 V)运行堆积胶;使用较高电压(140 V)运行分离胶(图5a和5b)。
5、运行约1小时,或直到染料前沿跑到凝胶底部(图6)。

图3. 向电泳槽中加入运行缓冲液。

图4. 移除梳子后,向凝胶中添加样品和分子标记物。

图5. (a) 样品通过堆积胶(低电压)运行;
(b) 样品通过分离胶(高电压)运行。

图6:将凝胶运行至电泳槽底部。
06 电转移
1、切割6张滤纸,使其大小适应凝胶的尺寸,并准备一张与凝胶尺寸相同的聚偏二氟乙烯(PVDF)膜。
2、将海绵和滤纸浸泡在转移缓冲液中,并将PVDF膜浸泡在甲醇中。
3、分开玻璃板,取出凝胶。
4、按以下顺序组装转移三明治:
海绵
3张滤纸
凝胶
PVDF膜
3张滤纸
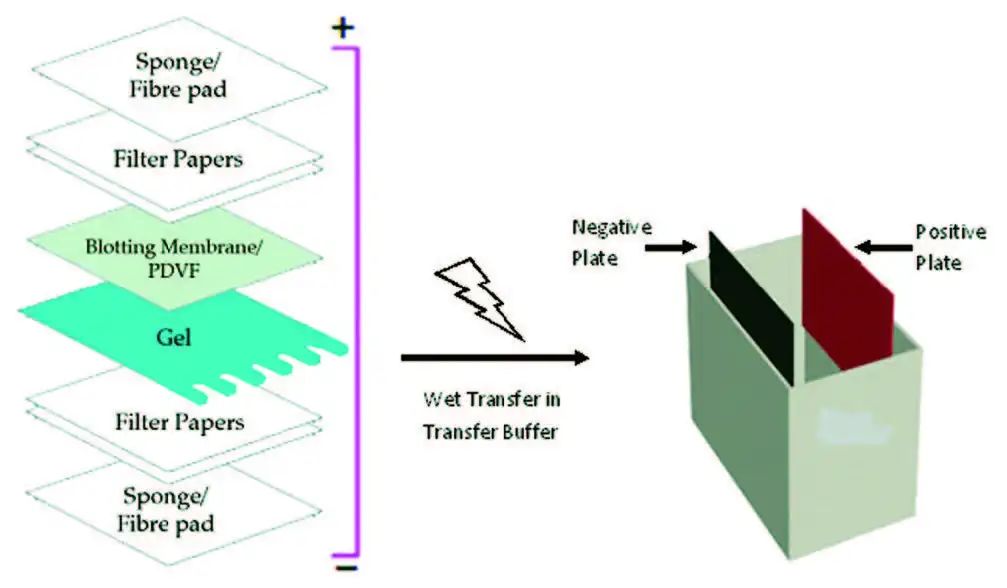
提示:确保凝胶和PVDF膜之间没有气泡,并挤出多余的液体。
5、将三明治移至转移装置中,设备应放在冰上以保持4ºC。向设备中添加转移缓冲液,确保三明治完全被缓冲液覆盖。将电极放在三明治上方,确保PVDF膜位于凝胶和正极之间(图7)。
6、转移约90分钟(图8)。
提示:转移时间应与凝胶的厚度成正比,因此对于0.75 mm厚的凝胶,转移时间可缩短为45分钟。

图7:转移应在冰上进行。
图8:转移后的膜。
07 封闭与抗体孵育
1、使用5%的脱脂奶粉溶液在TBST中封闭膜,封闭时间为1小时。
2、将一抗(主抗体)加入5%的牛血清白蛋白(BSA)中,在4°C摇床上孵育过夜(图9)。
3、使用TBST洗膜5分钟,重复洗涤3次。
提示:所有洗涤和抗体孵育步骤应在室温下使用摇床进行,以确保均匀震荡。
4、将二抗(次抗体)加入5%的脱脂奶粉溶液中,继续孵育1小时。
5、使用TBST洗膜5分钟,重复洗涤3次。
6、准备ECL混合液(按照生产商提供的A液和B液比例)。将膜浸泡在ECL溶液中1-2分钟(图10)。
提示:使用1000 µL移液管,确保ECL溶液完全覆盖膜的上下表面。
7、在黑暗室中进行结果显影(图11)。
提示:如果背景太强,减少曝光时间。
配方(TBST缓冲液)
1、将以下试剂溶解在800 mL蒸馏水中:
8.8 g NaCl
0.2 g KCl
3 g Tris
2、加入500 µL Tween-20
3、调整pH至7.4
4、用蒸馏水稀释至1 L
5、通过过滤或高压蒸汽灭菌进行灭菌

图9:使用摇床孵育膜与抗体。
图10:使用1000 μL移液管将ECL混合液孵育在膜上,以帮助反应过程。

图11:使用盒子在暗室中显影膜。
08 常见问题及解决办法
尽管Western Blot的操作程序相对简单,但在实验中仍可能出现许多问题,导致结果异常。问题可以分为五类:
1.异常或意外的条带
异常条带可能是由于蛋白酶降解引起的,导致出现意外位置的条带。此时建议使用新鲜样品,并保持其冷藏,或者更换抗体。如果蛋白质出现在过高的位置,可以尝试重新加热样品,以帮助破坏四级结构。同样,模糊条带通常是由高电压或转移过程中气泡引起的。此时应确保凝胶在较低电压下运行,并正确准备转移三明治。
更换运行缓冲液也能帮助解决此问题。非平坦条带可能是由于凝胶内的低电阻导致样品迁移过快。为解决这一问题,应优化凝胶以适应样品。最后,膜上的白色(负)条带是由于蛋白或抗体过量。
2.没有条带
没有条带的原因可能有很多,涉及抗体、抗原或使用的缓冲液。如果使用了不合适的抗体(无论是主抗体还是二抗),条带将不会显现。
抗体的浓度也很重要;如果抗体浓度太低,信号可能无法显示。需要注意的是,有些抗体不适用于Western Blot。没有条带的另一个原因可能是抗原浓度过低或缺失。此时,可以使用来自其他来源的抗原来确认问题是否出在样品或其他因素,如抗体。
过长时间的洗涤也可能降低信号。缓冲液也可能导致问题。应确保转移缓冲液、TBST、运行缓冲液和ECL都是新鲜且未污染的。如果缓冲液中污染了叠氮化钠,它可能会抑制HRP的活性。
3.信号弱
信号弱可能是由于抗体或抗原的浓度过低引起的。增加曝光时间也有助于增强条带的显现。
另一个可能的原因是脱脂奶粉掩盖了抗原。在这种情况下,可以使用BSA或减少奶粉的使用量。
4.背景过高
背景过高通常是由于抗体浓度过高,导致抗体与PVDF膜结合。另一个可能的问题是缓冲液过期。增加洗涤时间有助于减少背景。
过长时间的曝光也可能导致背景过高。因此,建议检查不同的曝光时间,以确定最佳曝光时间。
5.条带上的斑驳或不均匀
条带上的斑驳或不均匀通常是由于转移不当引起的。如果凝胶和膜之间有气泡,它会在胶片上显得较暗。使用摇床进行孵育可以避免孵育过程中出现不均匀的震荡。洗涤同样是非常重要的,它能帮助清洗背景。
这个问题也可能是由抗体与封闭剂结合引起的;在这种情况下,可以尝试更换封闭剂。过滤封闭剂也有助于去除一些杂质。最后,这个问题可能是由于二抗的聚集引起的;在这种情况下,应对二抗进行离心和过滤,以去除聚集体。
参考文献:Mahmood T, Yang P. Western blot: Technique,theory, and trouble shooting. North Am J Med Sci 2012;4:429-34.

