CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化

CUSTOMER
客户中心
助力生命健康领域从基础研究到产业化的加速转化
01 材料
1、实验动物
新生24h内的SPF级SD大鼠,体质量(6.0±0.5)g,雌雄不限。
2、药品及主要试剂
DMEM/F12(1:1)培养基和胎牛血清(FBS)、Neurobasal培养基、B27培养基添加剂、GlutaMAXsupplement(100X)、0.25 %胰酶(含EDTA、酚红)、青链霉素,小鼠抗Tau-4单克隆抗体,兔抗MAP-2多克隆抗体,小鼠抗GFAP单克隆抗体,AlexaFluor®Green488-conjugategoatanti-mouseIgG(H+L)、AlexaFluor®Red594-conjugategoatanti-rabbitIgG(H+L)、DAPI,多聚赖氨酸(PDL),MTS试剂盒。
3、主要仪器
恒温CO₂细胞培养箱,超净工作台,倒置显微镜,荧光倒置显微镜,酶标仪,台式低温离心机。
4、主要溶液的配制
(1)D-hanks液(1L):KCl 0.4g、NaCl 8.0g、Na₂HPO₄·12H₂O 0.132g、KH₂PO₄ 0.06g、NaHCO₃ 0.35g,分别称取以上各种药品溶于1L ddH₂O中,调 pH 值为 7.0~7.2,无菌滤膜过滤,101.3kPa,120℃高压灭菌20min,储存于4℃冰箱备用。
(2)多聚赖氨酸(PDL)溶液:PDL粉末用D-hanks液配成0.1mg/mL浓度,无菌滤膜过滤。
(3) 完全培养基配制(100mL):DMEM/F12(1:1)培养基 89% FBS 10%,青链霉素 1%。
(4)维持培养基配制(100mL):Neurobasal Media 96%、B27 2%、青链霉素 1%、GlutaMAX 1%。
02 方法
1、 细胞培养板的预处理
12 孔塑料培养板放入细胞爬片,用 0.1mg/mL 浓度的 PDL 包被细胞爬片 1h 或 4°C 包被过夜,用前用灭菌好的ddH₂O 漂洗 3 次,D-hanks 溶液漂洗 1 次,超净台干后,备用。
2、细胞培养
(1)取材:取出生24h内的SD大鼠经75 %乙醇浸泡、消毒,直接断头取脑于D-hanks平皿中,冰上剪开脑皮和脊髓,钝性分离出脑组织,完毕后取出“月牙形”海马组织,仔细去除脑膜、血细胞,快速将组织块剪成1mm×1mm×1mm大小。
(2)消化和单细胞悬液配置0.25%胰蛋白酶,37℃孵育10min,每间隔5min轻摇培养皿一次,脑组织块变圆、边缘毛刺、组织松软即终止消化,用完全培养基终止消化,吸出胰酶液,离心沉淀,200g离心5min,用Neurobasal无血清维持培养基,制成单细胞悬液。
(3)细胞计数:0.4%台盼蓝染计数法,计细胞数。将稀释后的细胞悬液滴于血细胞计数板的计数池中,在400倍镜下计数四个中方格内的总细胞,观察并区分活细胞和死细胞。计算公式如下:细胞密度(个/mL)=四个大格的细胞数×4×104×稀释倍数。
(4)细胞接种:调整细胞悬液密度为3.5×104个/mL,用移液器将单细胞悬液接种到12孔细胞培养板中,每孔2mL,37℃,放入饱和湿度和5%CO2细胞培养箱中进行细胞培养。
(5)细胞换液:提前将维持培养液(Neurobasal 96%、B27 2%、GluMAx1%、青链霉素 1%)预热至37℃,细胞培养24h后,弃去原培养液,加入维持培养基继续培养。此后每3d换液维持培养。
3、神经元形态学观察
倒置显微镜观察海马神经元在不同时段的形态学变化并拍照,比较不同阶段的形态和突触形成。

图1 原代大鼠海马神经元在不同时间的细胞生长状态( × 200,比例尺= 50μm,n = 3)
4、神经元细胞活性检测
神经细胞活性检测采用MTS法,使用酶标仪在490nm波长下测光密度(OD值),OD值越高说明细胞活性越强。将10μL的MTS检测试剂加入到96孔板中,测定1、2、3、4……12d的神经细胞活性神经细胞活性。用酶标仪检测波长在490nm的吸光度值(OD值),OD值越高说明细胞活性越大。实验需重复3次,取5组孔,以OD值为纵坐标绘制海马神经元生长曲线。
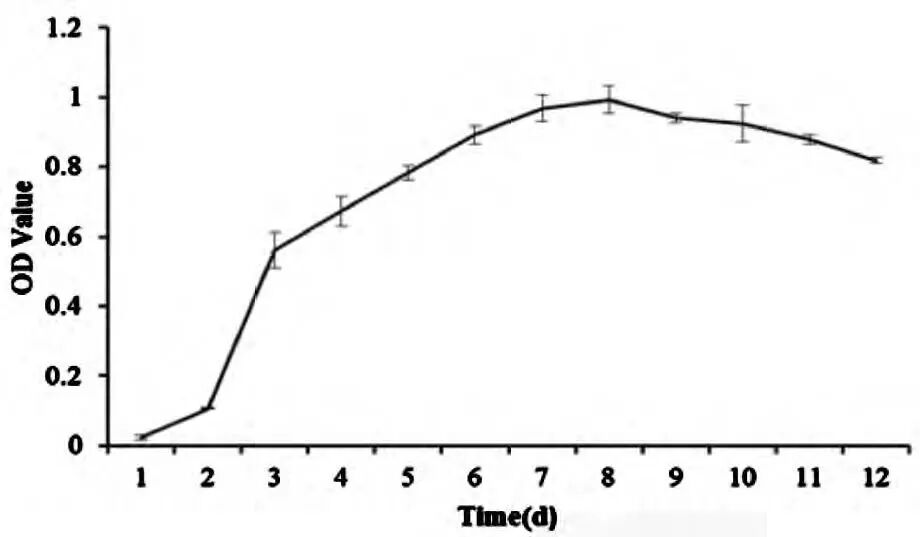
图2 海马神经元生长曲线图( n = 3,N = 5)
5、神经元纯度的鉴定
神经元培养第3d后弃培养液,将细胞用室温固定15min后以1% TritonX-100(PBS稀释)渗透15min后5% BSA封闭1h后滴加鼠抗β-III-tubulin(1:500稀释)或兔抗MAP-2抗体(1:500稀释),在4°C孵育一整夜。用PBS清洗后,滴加相应二抗(1:200稀释)37°C孵育1h(兔抗体用羊抗兔IgG,鼠抗体用羊抗鼠IgG)洗涤后,DAPI复染5min,PBS洗涤3次,避光封片后于荧光显微镜下观察。重复步骤同样进行GFAP抗体免疫荧光检测,检测胶质细胞污染情况。随机拍摄5个视野,计算神经元阳性细胞率。
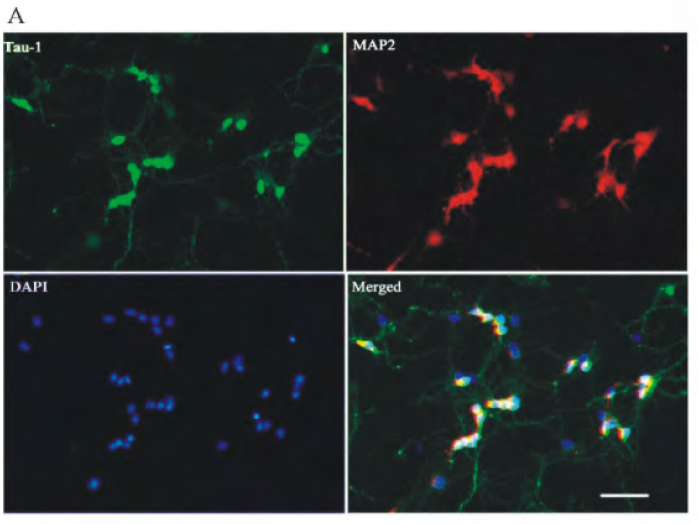
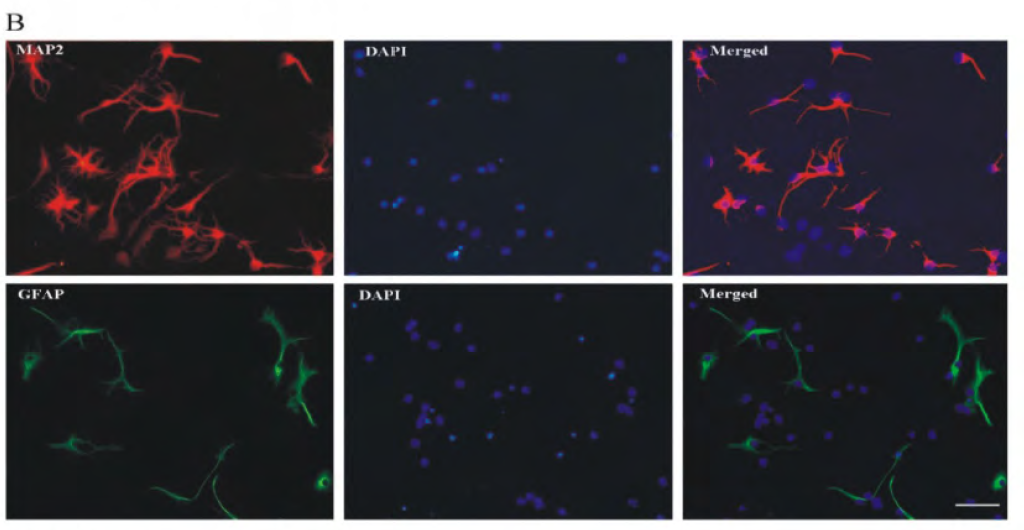
图3 海马神经元的鉴定(×100,Scale bar=50μm,n=3)
03 心得与经验
1、实验选取出生24h内新生鼠,可以不伤害孕鼠,节约资源,简便经济。且新生24h内新生鼠的脑组织方便固定,容易取材。
2、控制温度和时间:整个取材过程需要在冰袋上进行,并且所取海马组织置于冷的D-hanks液中,降低细胞代谢率,维持细胞充分的活力;同时取材过程速度要快,每只新生鼠操作时间控制在3~5min,总的时间控制在60min内,以防操作时间过长造成神经元失去活力甚至死亡。
3、剪碎海马组织时,需将其尽量快速剪碎呈1mm×1mm×1mm大小,以减少胰酶消化的时间。
4、胰酶消化组织时,注意控制胰酶的浓度和消化时间,将其放入37℃培养箱,先消化10min,轻轻摇晃组织,使其充分消化,然后弃掉胰酶,再加入新鲜的胰酶消化5min,以缩短胰酶消化时间并减少胰酶对细胞的损伤。
5、机械分离时注意吹打的力度和次数,尽量减少机械吹打对细胞造成的损伤,同时应尽量制成单细胞悬液,减少离心时间和转速。
6、神经元为贴壁培养细胞,必须粘附于固相表面才能生存,多聚赖氨酸包括左旋多聚赖氨酸(PLL)和右旋多聚赖氨酸(PDL),而PLL会损伤细胞,因此我们采用分子量为7~15万的PDL,包被细胞爬片和培养皿,以增加海马神经元的贴壁率。
7、接种前:充分混匀细胞,先接种200μL细胞悬液;接种后:放入培养箱培养3h后,每孔补加800μL无血清培养基继续培养。此方法既可以减少接种细胞液的用量,又可以缩短细胞至玻片的距离,使细胞更容易贴壁。
8、整个培养过程均采用无血清培养方法,未使用阿糖胞苷或5-氟尿嘧啶脱氧核苷,避免对神经元造成损伤。我们所用的Neurobasal无血清培养基为神经元专有培养,另外添加B27神经生长因子,可以选择性促进神经元生长,而抑制胶质细胞生长。
声明:本文使用图片来源于网络和文献,如有侵权请联系删除。

